
Insights into the RNA Virome of the Corn Leafhopper Dalbulus maidis, a Major Emergent Threat of Maize in Latin America




Abstract
1. Introduction
2. Material and Methods
2.1. Exploration of the D. maidis Virome from Public RNA-seq Datasets, Sequence Assembly, and Virus Identification
2.2. Bioinformatics Tools and Analyses
2.2.1. Sequence Analyses
2.2.2. Phylogenetic Analysis
3. Results and Discussion
3.1. RNA Viruses Associated with D. maidis
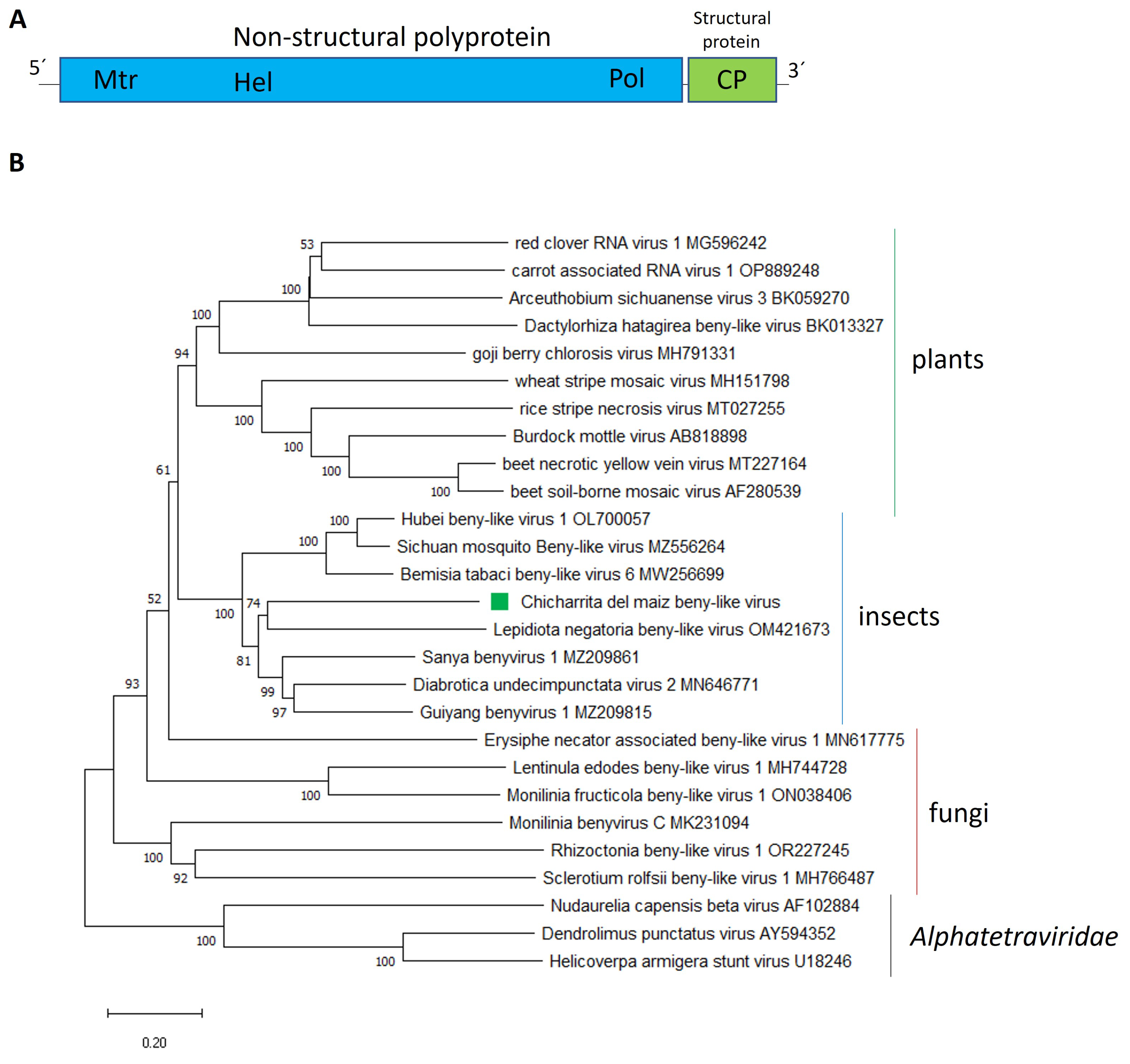
3.2. Molecular and Phylogenetic Characterization of the Newly Identified Maize Leafhopper Beny-Like Viruses
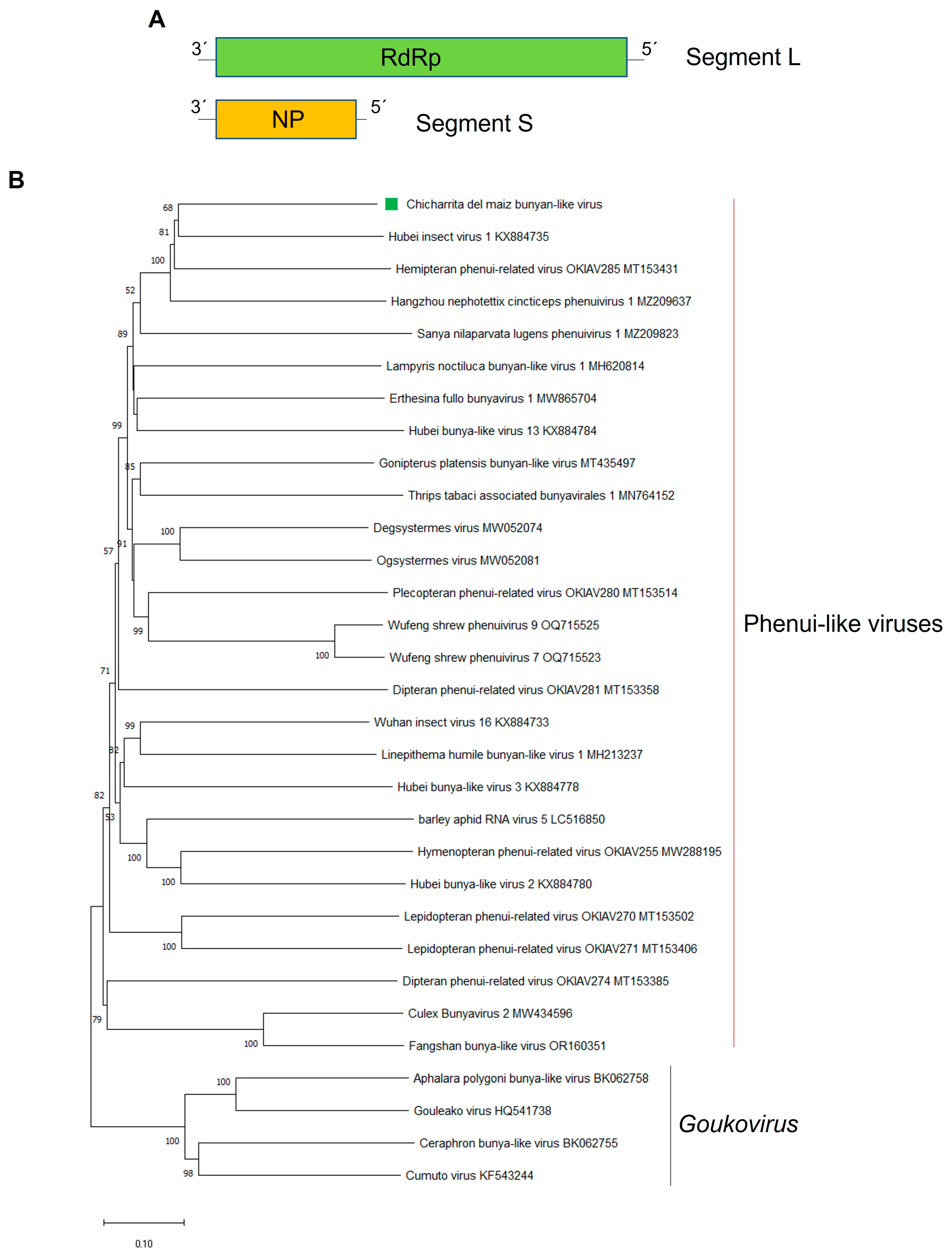
3.3. Molecular and Phylogenetic Characterization of the Newly Identified Maize Leafhopper Bunya-Like Virus
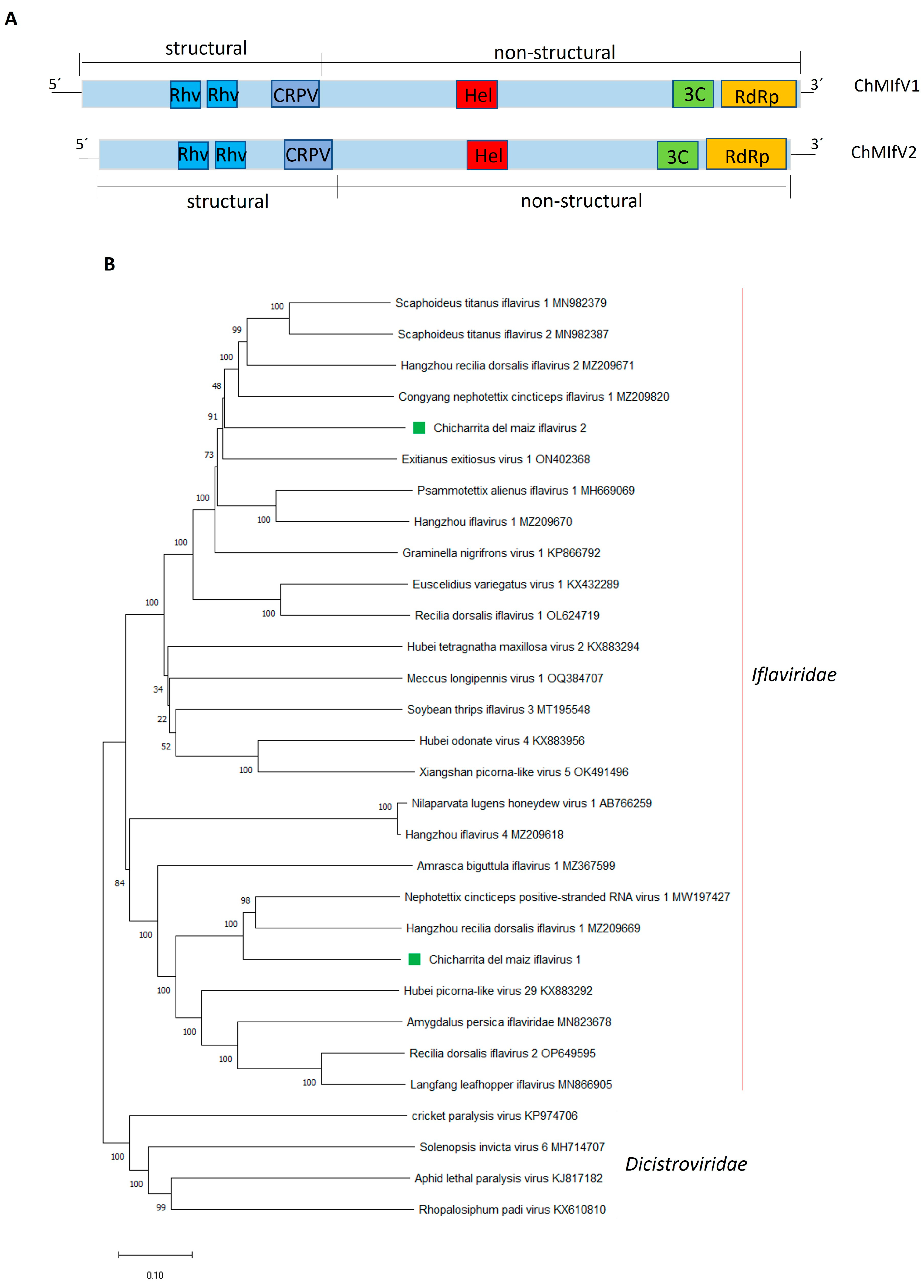
3.4. Molecular and Phylogenetic Characterization of the Newly Identified Maize Leafhopper Iflaviruses
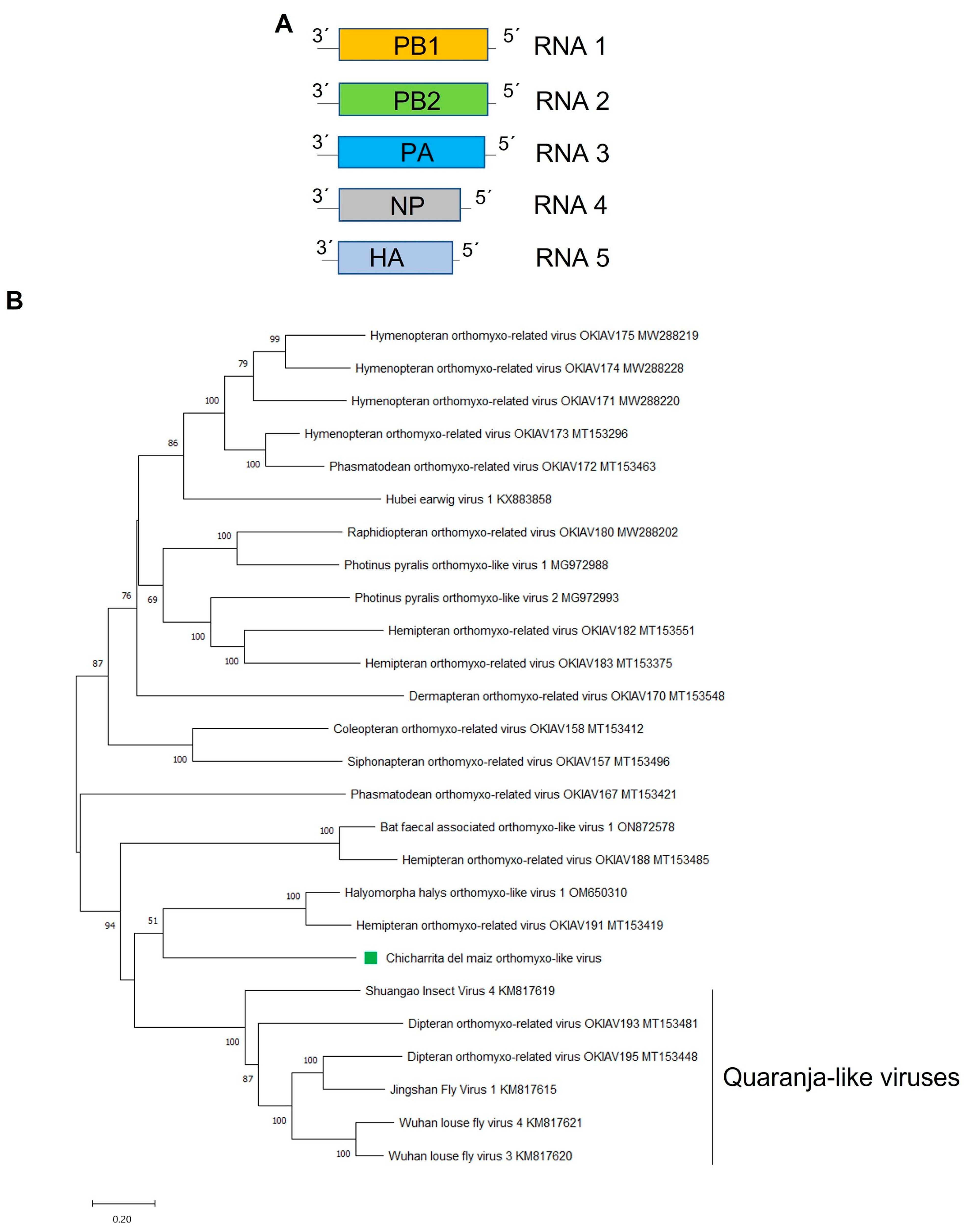
3.5. Molecular and Phylogenetic Characterization of the Newly Identified Maize Leafhopper Orthomyxo-Like Virus
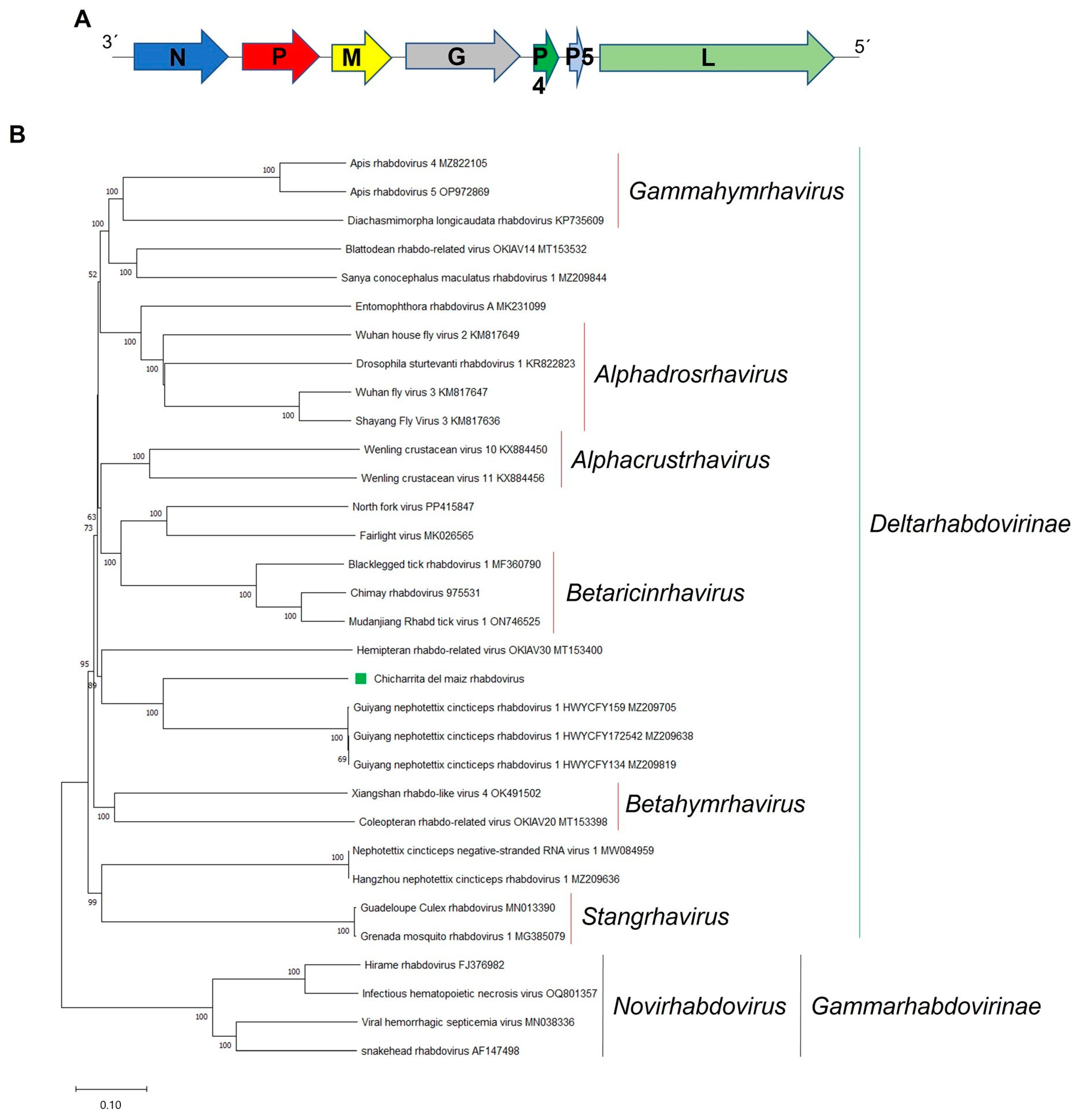
3.6. Molecular and Phylogenetic Characterization of the Newly Identified Maize Leafhopper Rhabdovirus
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nault, L.R. Maize bushy stunt and corn stunt: A comparison of disease symptoms, pathogen host ranges, and vectors. Phytopathology 1980, 70, 659–662. [Google Scholar] [CrossRef]
- Maluta, N.; Castro, T.; Spotti Lopes, J. DC-electrical penetration graph waveforms for Dalbulus maidis (Hemiptera: Cicadellidae) and the effects of entomopathogenic fungi on its probing behavior. Sci. Rep. 2023, 13, 22033. [Google Scholar] [CrossRef] [PubMed]
- Vilanova, E.S.; Ramos, A.; Souza de Oliveira, M.C.; Esteves, M.B.; Gonçalves, M.C.; Lopes, J.R.S. First report of a mastrevirus (Geminiviridae) transmitted by the corn leafhopper. Plant Dis. 2022, 106, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Nault, L.R. Evolution of an insect pest: Maize and the corn leafhopper, a case study. Maydica 1990, 35, 165–175. [Google Scholar]
- Pozebon, H.; Stürmer, G.R.; Arnemann, J.A. Corn stunt pathosystem and its leafhopper vector in Brazil. J. Econ. Entomol. 2022, 115, 1817–1833. [Google Scholar] [CrossRef] [PubMed]
- Carpane, P.; Catalano, M.I. Probing behavior of the corn leafhopper Dalbulus maidis on susceptible and resistant maize hybrids. PLoS ONE 2022, 17, e0259481. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Taylor, J.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Reche, I.; D’Orta, G.; Mladenov, N.; Winget, D.M.; Suttle, C.A. Deposition rates of viruses and bacteria above the atmosperic boundary layer. ISME J. 2018, 12, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Z.; Shi, M.; Holmes, E. Using metagenomics to characterize an expanding virosphere. Cell 2018, 172, 1168–1172. [Google Scholar] [CrossRef]
- Li, C.; Shi, M.; Tian, J.; Lin, X.; Kang, Y.; Chen, L.; Qin, X.; Xu, J.; Holmes, E.; Zhang, Y. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Käfer, S.; Paraskevopoulou, S.; Zirkel, F.; Wieseke, N.; Donath, A.; Petersen, M.; Jones, T.C.; Liu, S.; Zhou, X.; Middendorf, M.; et al. Re-assessing the diversity of negative strand RNA viruses in insects. PLoS Pathog. 2019, 15, e1008224. [Google Scholar] [CrossRef] [PubMed]
- Nouri, S.; Matsumura, E.E.; Kuo, Y.W.; Falk, B.W. Insect-Specific Viruses: From Discovery to Potential Translational Applications. Curr. Opin. Virol. 2018, 33, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.-H.; Ye, Z.-X.; Zhang, C.-X.; Chen, J.-P.; Li, J.-M. Diversity of RNA viruses in agricultural insects. Comput. Struct. Biotechnol. J. 2023, 21, 4312–4321. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pang, R.; Cheng, T.; Xue, L.; Zeng, H.; Lei, T.; Chen, M.; Wu, S.; Ding, Y.; Zhang, J.; et al. Abundant and Diverse RNA Viruses in Insects Revealed by RNA-Seq Analysis: Ecological and Evolutionary Implications. mSystems 2020, 5, e00039-20. [Google Scholar] [CrossRef] [PubMed]
- Harvey, E.; Holmes, E.C. Diversity and evolution of the animal virome. Nat. Rev. Microbiol. 2022, 20, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.; Zhang, X.; Vaas, J.; Klingler, F.; Mutz, P.; Dubin, A.; Pietschmann, T.; Roth, O.; Neuman, B.W.; Gorbalenya, A.E.; et al. Deep mining of the Sequence Read Archive reveals major genetic innovations in coronaviruses and other nidoviruses of aquatic vertebrates. PLoS Pathog. 2024, 20, e1012163. [Google Scholar] [CrossRef] [PubMed]
- Neri, U.; Wolf, Y.I.; Roux, S.; Camargo, A.P.; Lee, B.; Kazlauskas, D.; Chen, I.M.; Ivanova, N.; Allen, L.Z.; Paez-Espino, D.; et al. Expansion of the global RNA virome reveals diverse clades of bacteriophages. Cell 2022, 185, 4023–4037.e18. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, S.; Ka, S.; Zirkel, F.; Donath, A.; Petersen, M. Viromics of extant insect orders unveil the evolution of the flavi-like superfamily. Virus Evol. 2021, 7, veab030. [Google Scholar] [CrossRef]
- Rutar, S.O.; Kordis, D. Analysis of the RNA virome of basal hexapods. PeerJ 2020, 8, e8336. [Google Scholar] [CrossRef]
- Charon, J.; Olendraite, I.; Forgia, M.; Chong, L.C.; Hillary, L.S.; Roux, S.; Kupczok, A.; Debat, H.; Sakaguchi, S.; Tahzima, R.; et al. Consensus statement from the first RdRp Summit: Advancing RNA virus discovery at scale across communities. Front. Virol. 2024, 4, 1371958. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global organization and proposed megataxonomy of the virus world. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Wolf, Y.I.; Krupovic, M.; Zhang, Y.-Z.; Maes, P.; Dolja, V.V.; Koonin, E.V. Classify viruses—The gain is worth the pain. Nature 2019, 566, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Siddell, S.G.; Smith, D.B.; Adriaenssens, E.; Alfenas-Zerbini, P.; Dutilh, B.E.; Garcia, M.L.; Junglen, S.; Krupovic, M.; Kuhn, J.H.; Lambert, A.J.; et al. Virus taxonomy and the role of the International Committee on Taxonomy of Viruses (ICTV). J. Gen. Virol. 2023, 104, 001840. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus taxonomy in the age of metagenomics. Nature Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adriaenssens, E.M.; Zerbini, F.M.; Abrescia, N.G.A.; Aiewsakun, P.; Alfenas-Zerbini, P.; Bao, Y.; Barylski, J.; Drosten, C.; Duffy, S.; et al. Four principles to establish a universal virus taxonomy. PLoS Biol. 2023, 21, e3001922. [Google Scholar] [CrossRef] [PubMed]
- Bejerman, N.; Dietzgen, R.; Debat, H. Novel Tri-Segmented Rhabdoviruses: A Data Mining Expedition Unveils the Cryptic Diversity of cytorhabdoviruses. Viruses 2023, 15, 2402. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Stork, N.E.; NE, S. How Many Species of Insects and Other Terrestrial Arthropods Are There on Earth? Annu. Rev. Entomol. 2018, 63, 31–45. [Google Scholar] [CrossRef]
- Longdon, B.; Murray, G.G.R.; Palmer, W.J.; Day, J.P.; Parker, D.J.; Welch, J.J.; Obbard, D.; Jiggins, F.M. The evolution, diversity, and host associations of rhabdoviruses. Virus Evol. 2015, 1, vev014. [Google Scholar] [CrossRef]
- Bonning, B.C. The Insect Virome: Opportunities and Challenges. Curr. Issues Mol. Biol. 2020, 34, 1–12. [Google Scholar] [CrossRef]
- Jia, W.; Wang, F.; Xiao, S.; Yang, Y.; Chen, L.; Li, J.; Bao, Y.; Song, Q.; Ye, G. Identification and characterization of a novel rhabdovirus in green rice leafhopper, Nephotettix cincticeps. Virus Res. 2021, 296, 198281. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Y.; Liu, W.; Cao, M.; Wang, X. Sequence analysis and genomic organization of a novel chuvirus, Tàiyuán leafhopper virus. Arch. Virol. 2019, 164, 617–620. [Google Scholar] [CrossRef]
- Fu, Y.; Cao, M.; Wang, H.; Du, Z.; Liu, Y.; Li, J.; Bao, Y.; Song, Q.; Ye, G. Discovery and characterization of a novel insect-specific reovirus isolated from Psammotettix alienus. J. Gen. Virol. 2020, 101, 884–892. [Google Scholar] [CrossRef]
- Li, L.L.; Ye, Z.X.; Chen, J.P.; Zhang, C.X.; Huang, H.J.; Li, J.-M. Characterization of Two Novel Insect-Specific Viruses Discovered in the Green Leafhopper, Cicadella viridis. Insects 2022, 13, 378. [Google Scholar] [CrossRef] [PubMed]
- Ottati, S.; Chiapello, M.; Galetto, L.; Bosco, D.; Marzachì, C.; Abbà, S. New viral sequences identified in the Flavescence Dorée phytoplasma vector Scaphoideus titanus. Viruses 2020, 12, 287. [Google Scholar] [CrossRef]
- Gilmer, D.; Ratti, C.; Consortium, I.R. ICTV virus taxonomy profile: Benyviridae. J. Gen. Virol. 2017, 98, 1571–1572. [Google Scholar] [CrossRef]
- Etebari, K.; Lenancker, P.; Powell, K.S.; Furlong, M.J. Transcriptomics Reveal Several Novel Viruses from Canegrubs (Coleoptera: Scarabaeidae) in Central Queensland, Australia. Viruses 2022, 14, 649. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.J.; Ye, Z.X.; Wang, X.; Yan, X.T.; Zhang, Y.; He, Y.-J.; Qi, Y.-H.; Zhang, X.-D.; Zhuo, J.-C.; Lu, G.; et al. Diversity and infectivity of the RNA virome among different cryptic species of an agriculturally important insect vector: Whitefly Bemisia tabaci. NPJ Biofilms Microbiomes 2021, 7, 43. [Google Scholar] [CrossRef]
- Kondo, H.; Hirano, S.; Chiba, S.; Andika, I.B.; Hirai, M.; Maeda, T.; Tamada, T. Characterization of burdock mottle virus, a novel member of the genus Benyvirus, and the identification of benyvirus-related sequences in the plant and insect genomes. Virus Res. 2013, 177, 75–86. [Google Scholar] [CrossRef]
- Solovyev, A.G.; Morozov, S.Y. Uncovering Plant Virus Species Forming Novel Provisional Taxonomic Units Related to the family Benyviridae. Viruses 2022, 14, 2680. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Valencia-Jiménez, A.; Darlington, M.; Vélez, A.M.; Bonning, B.C. Diabrotica undecimpunctata virus 2, a Novel Small RNA Virus Discovered from Southern Corn Rootworm, Diabrotica undecimpunctata howardi Barber (Coleoptera: Chrysomelidae). Microbiol. Resour. Announc. 2020, 9, e00380-20. [Google Scholar] [CrossRef] [PubMed]
- Leventhal, S.S.; Wilson, D.; Feldmann, H.; Hawman, D.W. A Look into Bunyavirales Genomes: Functions of Non-Structural (NS) Proteins. Viruses 2021, 13, 314. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E. ICTV virus taxonomy profile: Iflaviridae. J. Gen. Virol. 2017, 98, 527–528. [Google Scholar] [CrossRef] [PubMed]
- van Oers, M. Genomics and biology of Iflaviruses. In Insect Virology; Asgari, S., Johnson, K., Eds.; Caister Academic Press: Norfolk, UK, 2010; pp. 231–250. [Google Scholar]
- Daveu, R.; Hervet, C.; Sigrist, L.; Sassera, D.; Jex, A.; Labadie, K.; Aury, J.-M.; Plantard, O.; Rispe, C. Sequence diversity and evolution of a group of iflaviruses associated with ticks. Arch. Virol. 2021, 166, 1843–1852. [Google Scholar] [CrossRef]
- Jia, W.; Wang, F.; Li, J.; Chang, X.; Yang, Y.; Yao, H.; Bao, Y.; Song, Q.; Ye, G. A Novel Iflavirus Was Discovered in Green Rice Leafhopper. Front. Microbiol. 2020, 11, 621141. [Google Scholar]
- Geng, P.; Li, W.; de Miranda, J.R.; Qian, Z.; An, L.; Terenius, O. Studies on the transmission and tissue distribution of Antheraea pernyi iflavirus in the Chinese oak silkmoth Antheraea pernyi. Virology 2017, 502, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, G.; de Miranda, J.R.; Boniotti, M.B.; Cameron, C.E.; Lavazza, A.; Capucci, L.; Camazine, S.M.; Rossi, C. Molecular and biological characterization of deformed wing virus of honeybees (Apis mellifera L.). J. Virol. 2006, 80, 4998–5009. [Google Scholar] [CrossRef] [PubMed]
- Wells, T.; Wolf, S.; Nicholls, E.; Groll, H.; Lim, K.S.; Clark, S.J.; Swain, J.; Osborne, J.L.; Haughton, A.J. Flight performance of actively foraging honey bees is reduced by a common pathogen. Environ. Microbiol. Rep. 2016, 8, 728–737. [Google Scholar] [CrossRef]
- Carballo, A.; Murillo, R.; Jakubowska, A.; Herrero, S.; Williams, T.; Caballero, P. Co-infection with iflaviruses influences the insecticidal properties of Spodoptera exigua multiple nucleopolyhedrovirus occlusion bodies: Implications for the production and biosecurity of baculovirus insecticides. PLoS ONE 2017, 12, e0177301. [Google Scholar] [CrossRef]
- Mengual-Martí, A.; Martínez-Solís, M.; Yousef-Yousef, M.; Garrido-Jurado, I.; Delgado-Sanfiel, P.; Quesada-Moraga, E.; Herrero, S. Impact of covert infections with an RNA virus on the susceptibility of Spodoptera exigua to natural enemies. BioControl 2022, 67, 605–615. [Google Scholar] [CrossRef]
- Ottati, S.; Persico, A.; Rossi, M.; Bosco, D.; Vallino, M.; Abbà, S.; Molinatto, G.; Palmano, S.; Balestrini, R.; Galetto, L.; et al. Biological characterization of Euscelidius variegatus iflavirus 1. J. Invertebr. Pathol. 2020, 173, 107370. [Google Scholar] [CrossRef]
- Shaw, M.L.; Palese, P. Orthomyxoviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2013; pp. 1151–1185. [Google Scholar]
- Papa, G.; Abbà, S.; Galetto, L.; Parise, C.; Marzachì, C.; Negri, I. Distribution and prevalence of viral genomes in Italian populations of the invasive brown marmorated stink bug Halyomorpha halys. J. Invertebr. Pathol. 2023, 200, 107977. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.; Murota, K.; Faizah, A.N.; Amoa-Bosompem, M.; Higa, Y.; Hayashi, T.; Tsuda, Y.; Sawabe, K.; Isawa, H. RNA virome analysis of hematophagous Chironomoidea flies (Diptera: Ceratopogonidae and Simuliidae) collected in Tokyo, Japan. Med. Entomol. Zool. 2020, 71, 225–243. [Google Scholar] [CrossRef]
- Walker, P.J.; Freitas-Astúa, J.; Bejerman, N.; Blasdell, K.R.; Breyta, R.; Dietzgen, R.G.; Fooks, A.R.; Kondo, H.; Kurath, G.; Kuzmin, I.V.; et al. ICTV Virus Taxonomy Profile: Rhabdoviridae 2022. J. Gen. Virol. 2022, 103, 001689. [Google Scholar] [CrossRef] [PubMed]
- Dietzgen, R.G.; Kondo, H.; Goodin, M.M.; Kurath, G.; Vasilakis, N. The family Rhabdoviridae: Mono- and bi-partite negative-sense RNA viruses with diverse genome organization and common evolutionary origins. Virus Res. 2017, 227, 158–170. [Google Scholar] [CrossRef]
- Schroeder, L.; Mar, T.B.; Haynes, J.R.; Wang, R.; Wempe, L.; Goodin, M.M. Host range and population survey of Spodoptera frugiperda rhabdovirus. J. Virol. 2019, 93, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Glodowski, D.R.; Petersen, J.M.; Dahlberg, J.E. Complex nuclear localization signals in the matrix protein of vesicular stomatitis virus. J. Biol. Chem. 2002, 277, 46864–46870. [Google Scholar] [CrossRef]
- Barrett, C.T.; Dutch, R.E. Viral Membrane Fusion and the Transmembrane Domain. Viruses 2020, 12, 693. [Google Scholar] [CrossRef]
- Walker, P.J.; Dietzgen, R.G.; Joubert, D.A.; Blasdell, K. Rhabdovirus accessory genes. Virus Res. 2011, 162, 110–125. [Google Scholar] [CrossRef]
- Walker, P.J.; Firth, C.; Widen, S.G.; Blasdell, K.R.; Guzman, H.; Wood, T.G.; Paradkar, P.N.; Holmes, E.C.; Tesh, R.B.; Vasilakis, N. Evolution of Genome Size and Complexity in the Rhabdoviridae. PLoS Pathog. 2015, 11, e1004664. [Google Scholar] [CrossRef]
- Joubert, D.A.; Blasdell, K.R.; Audsley, M.D.; Trinidad, L.; Monaghan, P.; Dave, K.A.; Lieu, K.G.; Amos-Ritchie, R.; Jans, D.A.; Moseley, G.W.; et al. Bovine Ephemeral Fever Rhabdovirus 1 Protein Has Viroporin-Like Properties and Binds Importin 1 and Importin 7. J. Virol. 2013, 88, 1591–1603. [Google Scholar] [CrossRef]
- Xia, X.; Cheng, A.; Wang, M.; Ou, X.; Sun, D.; Mao, S.; Huang, J.; Yang, Q.; Wu, Y.; Chen, S.; et al. Functions of Viroporins in the Viral Life Cycle and Their Regulation of Host Cell Responses. Front. Immunol. 2022, 13, 890549. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Name/Abbreviation | Accession Number | Genome/Segment Length (nt) | Protein ID/Length (aa) | Highest Scoring Virus-Protein/E-Value/Query Coverage%/Identity % (Blast P) |
|---|---|---|---|---|
| Chicharrita del maíz beny-like virus/ChMBLV | BK068250 | 6887 | Polyprotein/2003 structural P/224 | DuV2-polyprotein/0.0/70/46.61 DuV2-structural P/8 × 10−7/53/29.75 |
| Chicharrita del maíz bunyan-like virus/ChMBV | BK068254 BK068255 | Segment L 6651 Segment S 1281 | RdRp/2166 NP/327 | HbIV1-RdRp/0.0/99/49.84 HbIV1-NP/8 × 10−53/81/38.01 |
| Chicharrita del maíz iflavirus 1/ChMIfV1 | BK068251 | 11,104 | Polyprotein/3306 | NcPSRV1-polyprotein/0.0/96/64.03 |
| Chicharrita del maíz iflavirus 2/ChMIfV2 | BK068252 | 10,318 | Polyprotein/3102 | StIfV1-polyprotein/0.0/99/53.04 |
| Chicharrita del maíz orthomyxo-like virus/ChMOMLV | BK068245 BK068246 BK068247 BK068248 BK068249 | RNA1 2557 RNA2 2460 RNA3 2348 RNA4 1817 RNA5 1681 | PB1/794 PB2/777 PA/739 NP/572 HA/503 | HhOlV1-PB1/0.0/94/49.41 HhOlV1-PB2/4 × 10−82/98/26.32 BFaOMLV1-PA/4 × 10−123/99/34.59 HmOMRVOKIAV183-NP/3 × 10−60/89/29.87 Cotesiavirus orthomyxi-HA/2 × 10−20/73/24.56 |
| Chicharrita del maíz rhabdovirus/ChMRV | BK068253 | 12,271 * | N/454 P/311 M/242 G/662 U1/90 U2/52 L/2082 * | GuNCRV1-N/1 × 10−59/92/31.88 no hits GuNCRV1-M/3 × 10−28/74/29.83 GuNCRV1-G/7 × 10−144/95/38.79 no hits no hits GuNCRV1-L/0.0/99/48.44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Debat, H.; Farrher, E.S.; Bejerman, N. Insights into the RNA Virome of the Corn Leafhopper Dalbulus maidis, a Major Emergent Threat of Maize in Latin America. Viruses 2024, 16, 1583. https://doi.org/10.3390/v16101583
Debat H, Farrher ES, Bejerman N. Insights into the RNA Virome of the Corn Leafhopper Dalbulus maidis, a Major Emergent Threat of Maize in Latin America. Viruses. 2024; 16(10):1583. https://doi.org/10.3390/v16101583
Chicago/Turabian StyleDebat, Humberto, Esteban Simon Farrher, and Nicolas Bejerman. 2024. "Insights into the RNA Virome of the Corn Leafhopper Dalbulus maidis, a Major Emergent Threat of Maize in Latin America" Viruses 16, no. 10: 1583. https://doi.org/10.3390/v16101583
APA StyleDebat, H., Farrher, E. S., & Bejerman, N. (2024). Insights into the RNA Virome of the Corn Leafhopper Dalbulus maidis, a Major Emergent Threat of Maize in Latin America. Viruses, 16(10), 1583. https://doi.org/10.3390/v16101583