
Do NSm Virulence Factors in the Bunyavirales Viral Order Originate from Gn Gene Duplication?
 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
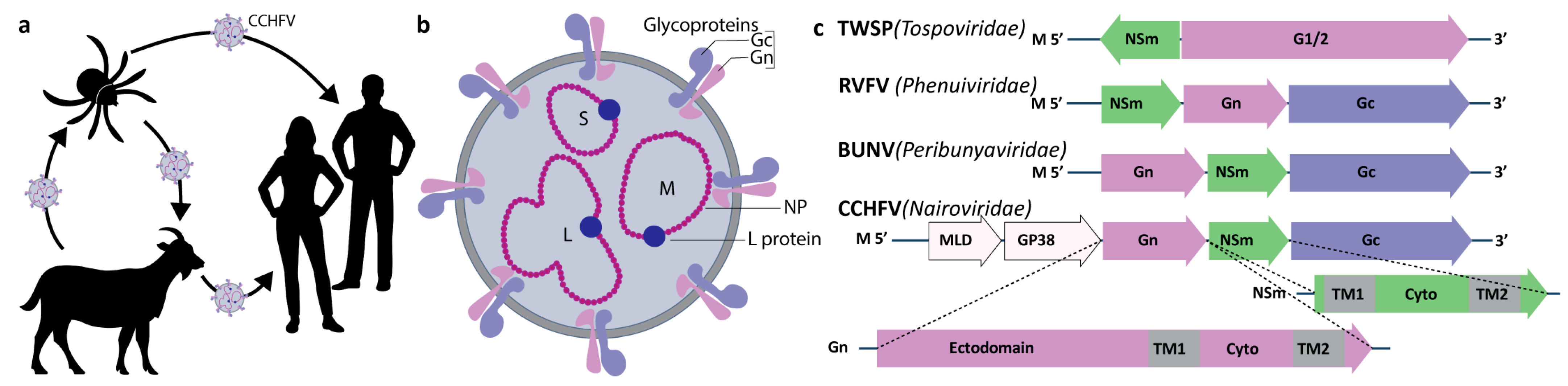
1. Introduction
2. Materials and Methods
3. Results
3.1. Full Conservation of the Gncyto Double Zinc Finger Was Predicted for the Nairoviridae NSm
3.2. NSm and Gncyto of the Peribunyaviridae Family Align Partially
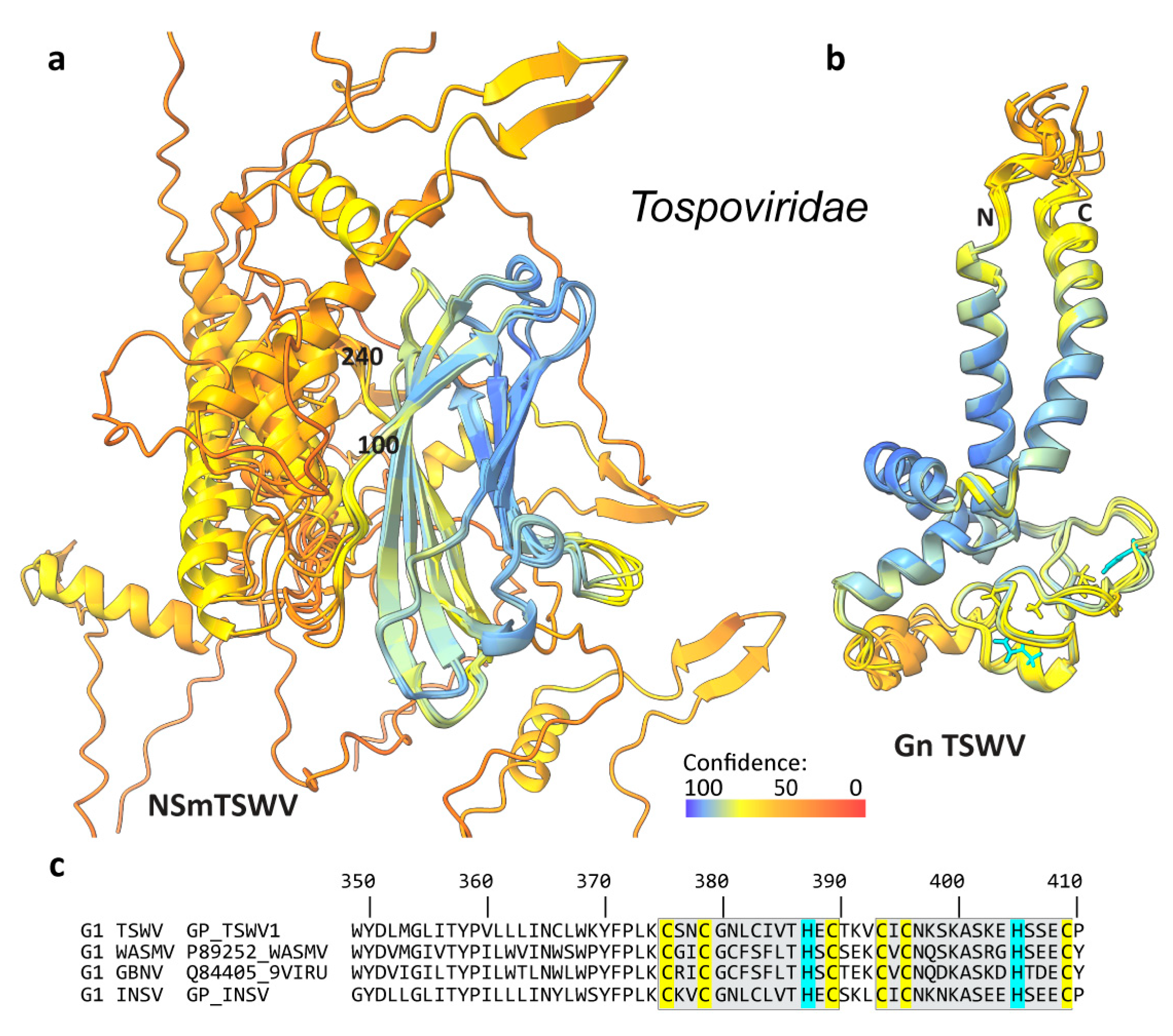
3.3. The NSm Proteins of the Phenuiviridae and Tospoviridae Families Were Predicted to Differ from Gncyto
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leventhal, S.S.; Wilson, D.; Feldmann, H.; Hawman, D.W. A Look into Bunyavirales Genomes: Functions of Non-Structural (NS) Proteins. Viruses 2021, 13, 314. [Google Scholar] [CrossRef]
- Ter Horst, S.; Conceição-Neto, N.; Neyts, J.; Rocha-Pereira, J. Structural and Functional Similarities in Bunyaviruses: Perspectives for Pan-bunya Antivirals. Rev. Med. Virol. 2019, 29, e2039. [Google Scholar] [CrossRef] [PubMed]
- Nakitare, G.W.; Elliott, R.M. Expression of the Bunyamwera Virus M Genome Segment and Intracellular Localization of NSm. Virology 1993, 195, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Altamura, L.A.; Bertolotti-Ciarlet, A.; Teigler, J.; Paragas, J.; Schmaljohn, C.S.; Doms, R.W. Identification of a Novel C-Terminal Cleavage of Crimean-Congo Hemorrhagic Fever Virus PreGN That Leads to Generation of an NSM Protein. J. Virol. 2007, 81, 6632–6642. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Botting, C.H.; Li, P.; Niglas, M.; Brennan, B.; Shirran, S.L.; Szemiel, A.M.; Elliott, R.M. Bunyamwera Orthobunyavirus Glycoprotein Precursor Is Processed by Cellular Signal Peptidase and Signal Peptide Peptidase. Proc. Natl. Acad. Sci. USA 2016, 113, 8825–8830. [Google Scholar] [CrossRef]
- Shi, X.; Kohl, A.; Léonard, V.H.J.; Li, P.; McLees, A.; Elliott, R.M. Requirement of the N-Terminal Region of Orthobunyavirus Nonstructural Protein NSm for Virus Assembly and Morphogenesis. J. Virol. 2006, 80, 8089–8099. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis. Protein Sci. 2017, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deléage, G. Gilbert NPS@: Network Protein Sequence Analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Estrada, D.F.; De Guzman, R.N. Structural Characterization of the Crimean-Congo Hemorrhagic Fever Virus Gn Tail Provides Insight into Virus Assembly. J. Biol. Chem. 2011, 286, 21678–21686. [Google Scholar] [CrossRef] [PubMed]
- Morellet, N.; Jullian, N.; De Rocquigny, H.; Maigret, B.; Darlix, J.L.; Roques, B.P. Determination of the Structure of the Nucleocapsid Protein NCp7 from the Human Immunodeficiency Virus Type 1 by 1H NMR. EMBO J. 1992, 11, 3059–3065. [Google Scholar] [CrossRef] [PubMed]
- Leastro, M.O.; Pallás, V.; Resende, R.O.; Sánchez-Navarro, J.A. The Movement Proteins (NSm) of Distinct Tospoviruses Peripherally Associate with Cellular Membranes and Interact with Homologous and Heterologous NSm and Nucleocapsid Proteins. Virology 2015, 478, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.S.; Martins, C.R.F.; Bezerra, I.C.; Nagata, T.; De Ávila, A.C.; Resende, R.O. Sequence Diversity of NS M Movement Protein of Tospoviruses. Arch. Virol. 2001, 146, 1267–1281. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin/Heidelberg, Germany, 1970; ISBN 978-3-642-86661-6. [Google Scholar]
- Lallemand, T.; Leduc, M.; Landès, C.; Rizzon, C.; Lerat, E. An Overview of Duplicated Gene Detection Methods: Why the Duplication Mechanism Has to Be Accounted for in Their Choice. Genes 2020, 11, 1046. [Google Scholar] [CrossRef] [PubMed]
- Cisneros-Martínez, A.M.; Becerra, A.; Lazcano, A. Ancient Gene Duplications in RNA Viruses Revealed by Protein Tertiary Structure Comparisons. Virus Evol. 2021, 7, veab019. [Google Scholar] [CrossRef]
- Mishra, A.K.; Moyer, C.L.; Abelson, D.M.; Deer, D.J.; El Omari, K.; Duman, R.; Lobel, L.; Lutwama, J.J.; Dye, J.M.; Wagner, A.; et al. Structure and Characterization of Crimean-Congo Hemorrhagic Fever Virus GP38. J. Virol. 2020, 94, e02005-19. [Google Scholar] [CrossRef] [PubMed]
- Hellert, J.; Aebischer, A.; Wernike, K.; Haouz, A.; Brocchi, E.; Reiche, S.; Guardado-Calvo, P.; Beer, M.; Rey, F.A. Orthobunyavirus Spike Architecture and Recognition by Neutralizing Antibodies. Nat. Commun. 2019, 10, 879. [Google Scholar] [CrossRef]
- Estrada, D.F.; Boudreaux, D.M.; Zhong, D.; St. Jeor, S.C.; De Guzman, R.N. The Hantavirus Glycoprotein G1 Tail Contains Dual CCHC-Type Classical Zinc Fingers. J. Biol. Chem. 2009, 284, 8654–8660. [Google Scholar] [CrossRef] [PubMed]
- Estrada, D.F.; Conner, M.; Jeor, S.C.; Guzman, R.N.D. The Structure of the Hantavirus Zinc Finger Domain Is Conserved and Represents the Only Natively Folded Region of the Gn Cytoplasmic Tail. Front. Microbio. 2011, 2, 251. [Google Scholar] [CrossRef] [PubMed]
- Bragantini, B.; Tiotiu, D.; Rothé, B.; Saliou, J.-M.; Marty, H.; Cianférani, S.; Charpentier, B.; Quinternet, M.; Manival, X. Functional and Structural Insights of the Zinc-Finger HIT Protein Family Members Involved in Box C/D snoRNP Biogenesis. J. Mol. Biol. 2016, 428, 2488–2506. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefebvre, V.; Leon Foun Lin, R.; Cole, L.; Cosset, F.-L.; Fogeron, M.-L.; Böckmann, A. Do NSm Virulence Factors in the Bunyavirales Viral Order Originate from Gn Gene Duplication? Viruses 2024, 16, 90. https://doi.org/10.3390/v16010090
Lefebvre V, Leon Foun Lin R, Cole L, Cosset F-L, Fogeron M-L, Böckmann A. Do NSm Virulence Factors in the Bunyavirales Viral Order Originate from Gn Gene Duplication? Viruses. 2024; 16(1):90. https://doi.org/10.3390/v16010090
Chicago/Turabian StyleLefebvre, Victor, Ravy Leon Foun Lin, Laura Cole, François-Loïc Cosset, Marie-Laure Fogeron, and Anja Böckmann. 2024. "Do NSm Virulence Factors in the Bunyavirales Viral Order Originate from Gn Gene Duplication?" Viruses 16, no. 1: 90. https://doi.org/10.3390/v16010090
APA StyleLefebvre, V., Leon Foun Lin, R., Cole, L., Cosset, F.-L., Fogeron, M.-L., & Böckmann, A. (2024). Do NSm Virulence Factors in the Bunyavirales Viral Order Originate from Gn Gene Duplication? Viruses, 16(1), 90. https://doi.org/10.3390/v16010090