
Antivirals for Broader Coverage against Human Coronaviruses



Abstract
1. Introduction
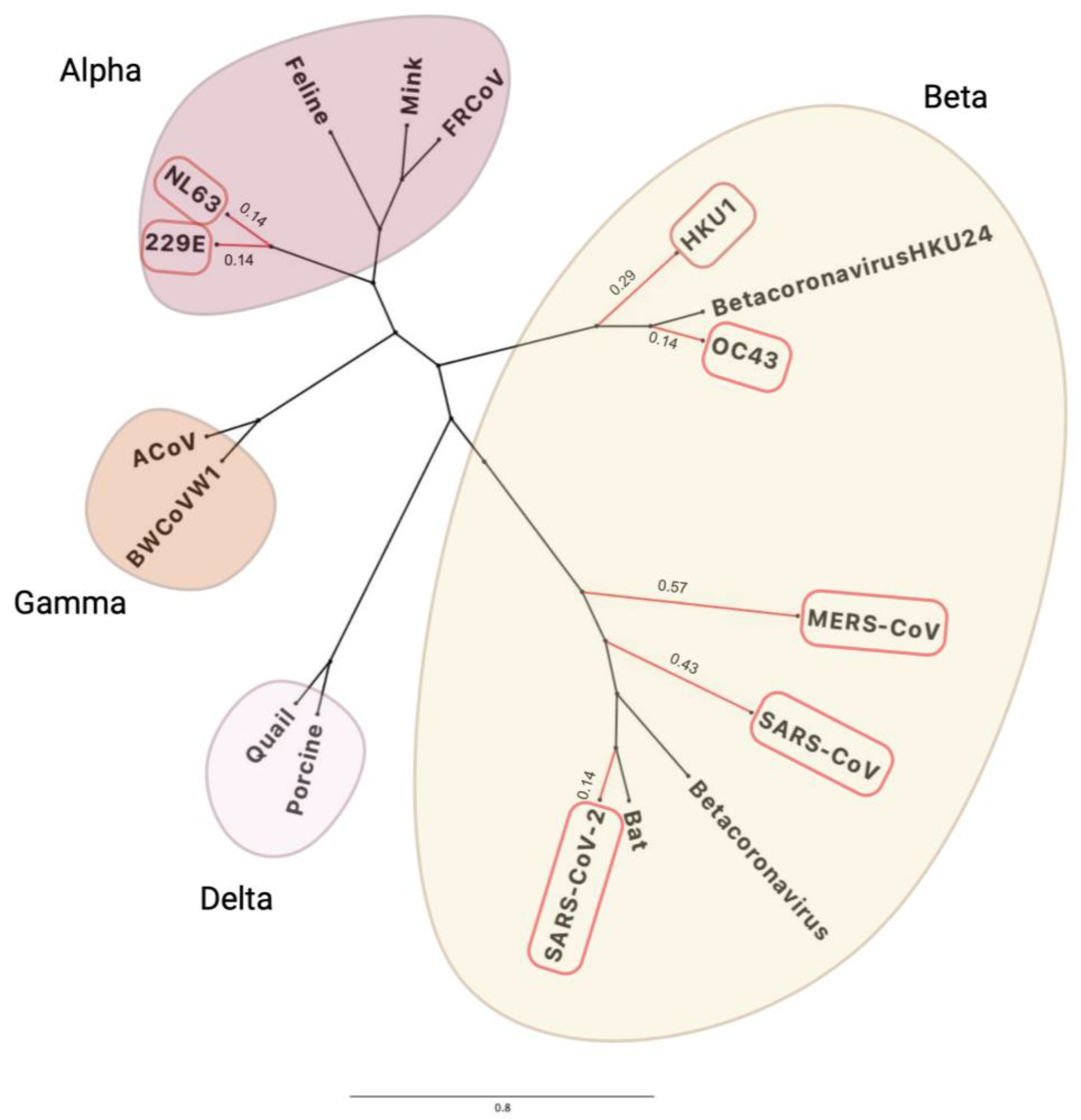
1.1. History of Human Coronaviruses
1.2. CoV Genes and Genetic Diversity

| Protein | Functions in SARS-CoV-2 | 3D Structure |
|---|---|---|
| NSP1 | ~180 a.a. promotes viral gene expression and inhibits immune functions [39]. | 7K7P |
| NSP2 | ~638 a.a. interacts with host factors prohibitin 1 and prohibitin 2, which are involved in many cellular processes including mitochondrial biogenesis [40]. | 7EXM (N terminus) |
| NSP3 | ~1945 a.a, papain-like protease (PLpro) processes the viral polyprotein [41]. | 6WOJ |
| NSP4 | ~500 a.a. transmembrane glycoprotein forms DMVs in complex with NSP3 [42]. | Not available |
| NSP5 | ~306 a.a, main protease (3CLpro/MPro) cleaves polyprotein release nsp4-nsp16 [43]. | 8DDI |
| NSP6 | ~290 a.a, complexes with NSP3 and NSP4 to induce DMVs in infected cells [44]. | 8DRS (complex with NSP6 and 7) |
| NSP7 | ~83 a.a. forms supercomplex with NSP8 and NSP12 to process viral RNA [45]. | 7DCD (complex with NSP8) |
| NSP8 | ~198 a.a forms supercomplex with NSP7 and NSP12 to process viral RNA [45]. | 7DCD (complex with NSP7) |
| NSP9 | ~113 a.a interacts with NSP12, unclear specific functions [46]. | 3EE7 |
| NSP10 | ~139 a.a. forms a dodecamer complex with NSP14 and NSP16 to stimulate 3′-5′ exoribonuclease and 2′-O-methyltransferase activities [47]. | 6W4H (complex with NSP16) |
| NSP11 | ~13–23 a.a pp1a cleavage product at the nsp10/11 boundary. Function unknown [48]. | Not available |
| NSP12 | ~932 a.a RdRp performing replication and transcription of the viral genome [49]. | 7BW4 |
| NSP13 | ~601 a.a the main helicase for the CoVs [50]. | 7NIO |
| NSP14 | ~527 a.a 3′-5′ exoribonuclease proofreading mechanism (ExoN) in complex with NSP10 and viral mRNA capping activities [51]. | 7QGI |
| NSP15 | ~346 a.a important for immune evasion by preventing dsRNA sensor activation [52]. | 7DW0 (complex with NSP5/14) |
| NSP16 | ~298 a.a 2′-O-methyltransferase activity, activated once in complex with NSP10 [53]. | 6W4H (complex with NSP10) |
| ORF3a | ~275 a.a viroporin iron channel in SARS-CoV, promotes viral movement, release and activation of inflammasomes [54]. | 6XDC |
| ORF3b | ~22 a.a interrupting interferon antagonistic functions, but not fully understood [55]. | Not available |
| ORF6 | ~61 a.a interferes with innate immune responses, suppressing kinases and types I and II IFN pathways [56]. | 7VPH (complex with Rae1-Nup98) |
| ORF7a | ~121 a.a type I membrane protein that interacts with CD14+ monocytes and increases glycosylation for immune evasion of presenting antigens [57]. | 7CI3 (ectodomain) |
| ORF7b | ~43 a.a interference with cellular processes and infection symptoms [57]. | Not available |
| ORF8 | ~121 a.a interferon antagonist to promoting cytokine storms [57]. | 7JTL |
| ORF9b | ~97 a.a. localised in mitochondrial membranes, associated with IFN responses [58]. | 7YE7 |
| ORF9c | ~70 a.a interacts with host proteins, involvement in ER stress responses and lipid remodelling [57]. | Not available |
| ORF10 | ~38 a.a not necessary for SARS-CoV-2 infection [57]. | Not available |
| Spike (S) (ORF2) | ~1273 a.a interacts with host entry receptors, facilitates fusion [59]. | 8C8P (complex with mAb). |
| Membrane (M) (ORF5) | ~222 a.a mediates assembly, packaging and budding of viral particles [60] | 8CTK |
| Envelope (E) (ORF4) | ~75 a.a involved in viral assembly, budding, and pathogenesis [61]. | 7K3G |
| Nucleocapsid (N) (ORF9a) | ~419 a.a. involved in genome protection, viral RNA replication, virion assembly, and immune evasion [62]. | 6WZO |
1.3. CoV Genetic Drift
| HCoV | Entry Receptors |
| CoV-NL63 | Angiotensin converting enzyme 2 (ACE-2) |
| CoV-229E | Aminopeptidase N (APN) |
| CoV-HKU1 | 9-O-acetylated sialic acids (9-O-Ac-Sias) |
| CoV-OC43 | 9-O-acetylated sialic acids (9-O-Ac-Sias) |
| SARS-CoV | Angiotensin converting enzyme 2 (ACE-2) |
| MERS-CoV | Dipeptidyl peptidase 4 (DPP4) |
| SARS-CoV-2 | Angiotensin converting enzyme 2 (ACE-2) |
2. Treatment of HCoV Infection
3. Novel Viral Druggable Targets
3.1. Non-Structural Protein 3 (NSP3)
3.2. Non-Structural Protein 5 (NSP5)
3.3. Non-Structural Protein 12 (NSP12)
4. Available and Alternative Treatments/Prophylaxis
- Paxlovid (ritonavir and nirmatrelvir);
- Lagevrio (molnupiravir);
- Veklury (remdesivir)—this is only approved for use in individuals who are at high risk of developing severe COVID-19 infection, including the vulnerable/immunocompromised population.
- Sotrovimab;
- Bebtelovimab;
- Casirivimab/imdevimab.
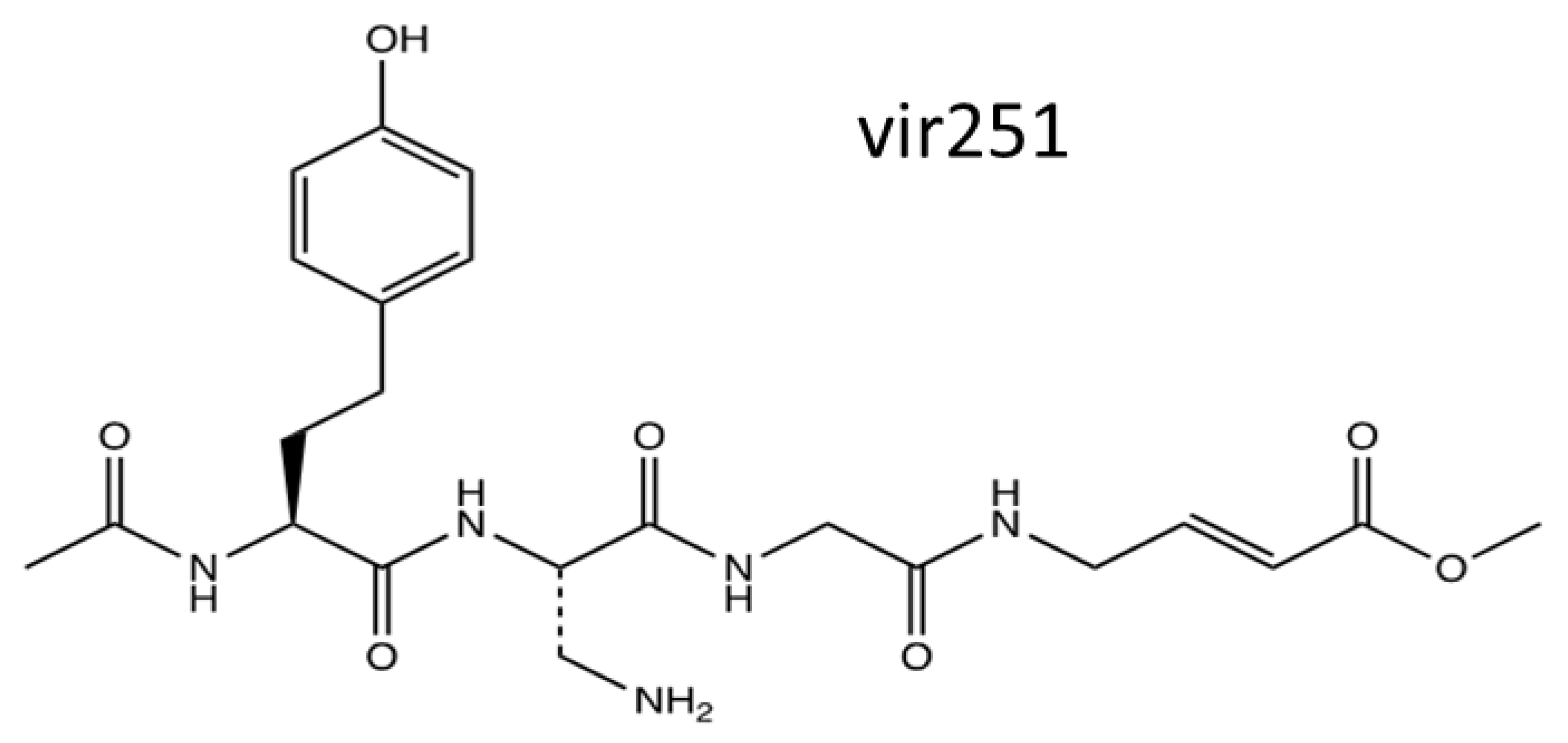
5. The Design of a Novel Antiviral
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Agarwal, R. The aftermath of coronavirus disease 2019: Devastation or a new dawn for nephrology? Nephrol. Dial. Transpl. 2020, 35, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). Encycl. Virol. 2021, 2, 428–440. [Google Scholar] [CrossRef]
- Ye, Z.-W.; Yuan, S.; Yuen, K.-S.; Fung, S.-Y.; Chan, C.-P.; Jin, D.-Y. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, M.; Hamblin, M.R.; Rezaei, N. COVID-19: Transmission, prevention, and potential therapeutic opportunities. Clin. Chim. Acta 2020, 508, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. COVID-19: New “Pirola” variant BA.2.86 continues to spread in UK and US. BMJ 2023, 382, p2097. [Google Scholar] [CrossRef]
- Hodgens, A.; Gupta, V. Severe Acute Respiratory Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK558977/ (accessed on 3 May 2022).
- Abdul-Rasool, S.; Fielding, B.C. Understanding Human Coronavirus HCoV-NL63. Open Virol. J. 2010, 4, 76–84. [Google Scholar] [CrossRef]
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK554776/ (accessed on 9 May 2023).
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 2020, 21, 224. [Google Scholar] [CrossRef]
- Abdelrahman, Z.; Li, M.; Wang, X. Comparative Review of SARS-CoV-2, SARS-CoV, MERS-CoV, and Influenza A Respiratory Viruses. Front. Immunol. 2020, 11, 2309. [Google Scholar] [CrossRef]
- Yang, Y.; Peng, F.; Wang, R.; Guan, K.; Jiang, T.; Xu, G.; Sun, J.; Chang, C. The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun. 2020, 109, 102434. [Google Scholar] [CrossRef]
- Azhar, E.I.; Hui, D.S.C.; Memish, Z.A.; Drosten, C.; Zumla, A. The Middle East Respiratory Syndrome (MERS). Infect. Dis. Clin. North. Am. 2019, 33, 891–905. [Google Scholar] [CrossRef]
- CSR. World Health Organization–Regional Office for the Eastern Mediterranean. MERS Outbreaks. Available online: http://www.emro.who.int/health-topics/mers-cov/mers-outbreaks.html (accessed on 26 July 2023).
- Barry, M.; Phan, M.V.T.; Akkielah, L.; Al-Majed, F.; Alhetheel, A.; Somily, A.; Alsubaie, S.S.; McNabb, S.J.N.; Cotten, M.; Zumla, A.; et al. Nosocomial outbreak of the Middle East Respiratory Syndrome coronavirus: A phylogenetic, epidemiological, clinical and infection control analysis. Travel. Med. Infect. Dis. 2020, 37, 101807. [Google Scholar] [CrossRef] [PubMed]
- Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Available online: https://www.who.int/news-room/fact-sheets/detail/middle-east-respiratory-syndrome-coronavirus-(mers-cov) (accessed on 22 November 2023).
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 8 May 2023).
- Zhou, H.; Yang, J.; Zhou, C.; Chen, B.; Fang, H.; Chen, S.; Zhang, X.; Wang, L.; Zhang, L. A Review of SARS-CoV2: Compared with SARS-CoV and MERS-CoV. Front. Med. 2021, 8, 628370. [Google Scholar] [CrossRef] [PubMed]
- Pustake, M.; Tambolkar, I.; Giri, P.; Gandhi, C. SARS, MERS and COVID-19: An overview and comparison of clinical, laboratory and radiological features. J. Fam. Med. Prim. Care 2022, 11, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Luk, H.K.H.; Wong, A.C.P.; Li, K.S.M.; Zhu, L.; He, Z.; Fung, J.; Chan, T.T.Y.; Fung, K.S.C.; Woo, P.C.Y. Possible Bat Origin of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Ghai, R.R.; Carpenter, A.; Liew, A.Y.; Martin, K.B.; Herring, M.K.; Gerber, S.I.; Hall, A.J.; Sleeman, J.M.; VonDobschuetz, S.; Behravesh, C.B. Animal Reservoirs and Hosts for Emerging Alphacoronaviruses and Betacoronaviruses. Emerg. Infect. Dis. 2021, 27, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A.; Kennedy, B.K. The World Goes Bats: Living Longer and Tolerating Viruses. Cell Metab. 2020, 32, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Baker, M.L.; Kulcsar, K.; Misra, V.; Plowright, R.; Mossman, K. Novel Insights Into Immune Systems of Bats. Front. Immunol. 2020, 11, 26. [Google Scholar] [CrossRef]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef]
- Kaur, N.; Singh, R.; Dar, Z.; Bijarnia, R.K.; Dhingra, N.; Kaur, T. Genetic comparison among various coronavirus strains for the identification of potential vaccine targets of SARS-CoV2. Infect. Genet. Evol. 2021, 89, 104490. [Google Scholar] [CrossRef]
- Mallapaty, S. Closest known relatives of virus behind COVID-19 found in Laos. Nature 2021, 597, 603. [Google Scholar] [CrossRef]
- Cyranoski, D. Bat cave solves mystery of deadly SARS virus—And suggests new outbreak could occur. Nature 2017, 552, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.A.; Sacco, O.; Mancino, E.; Cristiani, L.; Midulla, F. Differences and similarities between SARS-CoV and SARS-CoV-2: Spike receptor-binding domain recognition and host cell infection with support of cellular serine proteases. Infection 2020, 48, 665–669. [Google Scholar] [CrossRef] [PubMed]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 25 February 2022).
- Wang, N.; Shang, J.; Jiang, S.; Du, L. Subunit Vaccines Against Emerging Pathogenic Human Coronaviruses. Front. Microbiol. 2020, 11, 298. [Google Scholar] [CrossRef] [PubMed]
- Binet, M.; Gascuel, O.; Scornavacca, C.; Douzery, E.J.P.; Pardi, F. Fast and accurate branch lengths estimation for phylogenomic trees. BMC Bioinform. 2016, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Information NC for B, Pike USNL of M 8600 R, MD B, Usa 20894. National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 13 January 2022).
- Artic Network. Available online: https://artic.network/how-to-read-a-tree.html# (accessed on 26 July 2023).
- El-Sayed, A.; Kamel, M. Coronaviruses in humans and animals: The role of bats in viral evolution. Environ. Sci. Pollut. Res. Int. 2021, 28, 19589–19600. [Google Scholar] [CrossRef] [PubMed]
- Temmam, S.; Vongphayloth, K.; Baquero, E.; Munier, S.; Bonomi, M.; Regnault, B.; Douangboubpha, B.; Karami, Y.; Chrétien, D.; Sanamxay, D.; et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 2022, 604, 330–336. [Google Scholar] [CrossRef] [PubMed]
- BioRender. Available online: https://app.biorender.com/illustrations/627b7981d323fa4528ad7c00 (accessed on 11 May 2022).
- Ellis, P.; Somogyvári, F.; Virok, D.P.; Noseda, M.; McLean, G.R. Decoding Covid-19 with the SARS-CoV-2 Genome. Curr. Genet. Med. Rep. 2021, 9, 1–12. [Google Scholar] [CrossRef]
- Krichel, B.; Falke, S.; Hilgenfeld, R.; Redecke, L.; Uetrecht, C. Processing of the SARS-CoV pp1a/ab nsp7–10 region. Biochem. J. 2020, 477, 1009–1019. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Coronaviruses 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.-A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef]
- Cornillez-Ty, C.T.; Liao, L.; Yates, J.R.; Kuhn, P.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J. Virol. 2009, 83, 10314–10318. [Google Scholar] [CrossRef]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.P.; Almasy, K.M.; McDonald, E.F.; Plate, L. Comparative multiplexed interactomics of SARS-CoV-2 and homologous coronavirus non-structural proteins identifies unique and shared host-cell dependencies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Scott, B.M.; Lacasse, V.; Blom, D.G.; Tonner, P.D.; Blom, N.S. Predicted coronavirus Nsp5 protease cleavage sites in the human proteome. BMC Genom. Data 2022, 23, 25. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, Y.; Huang, Z.; Xu, W.; Hu, W.; Yi, L.; Liu, Z.; Chan, H.; Zeng, J.; Liu, X.; et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. Cell Death Differ. 2022, 29, 1240–1254. [Google Scholar] [CrossRef]
- Reshamwala, S.M.S.; Likhite, V.; Degani, M.S.; Deb, S.S.; Noronha, S.B. Mutations in SARS-CoV-2 nsp7 and nsp8 proteins and their predicted impact on replication/transcription complex structure. J. Med. Virol. 2021, 93, 4616–4619. [Google Scholar] [CrossRef]
- El-Kamand, S.; Du Plessis, M.-D.; Breen, N.; Johnson, L.; Beard, S.; Kwan, A.H.; Richard, D.J.; Cubeddu, L.; Gamsjaeger, R. A distinct ssDNA/RNA binding interface in the Nsp9 protein from SARS-CoV-2. Proteins Struct. Funct. Bioinform. 2022, 90, 176–185. [Google Scholar] [CrossRef]
- Lin, S.; Chen, H.; Chen, Z.; Yang, F.; Ye, F.; Zheng, Y.; Yang, J.; Lin, X.; Sun, H.; Wang, L.; et al. Crystal structure of SARS-CoV-2 nsp10 bound to nsp14-ExoN domain reveals an exoribonuclease with both structural and functional integrity. Nucleic Acids Res. 2021, 49, 5382–5392. [Google Scholar] [CrossRef]
- Gadhave, K.; Kumar, P.; Kumar, A.; Bhardwaj, T.; Garg, N.; Giri, R. Conformational dynamics of 13 amino acids long NSP11 of SARS-CoV-2 under membrane mimetics and different solvent conditions. Microb. Pathog. 2021, 158, 105041. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, Z.; Xiao, X.; Tian, Z.; Dong, X.; Wang, C.; Li, L.; Ren, L.; Lei, X.; Xiang, Z.; et al. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell Mol. Immunol. 2021, 18, 945–953. [Google Scholar] [CrossRef]
- Fung, S.-Y.; Siu, K.-L.; Lin, H.; Chan, C.-P.; Yeung, M.L.; Jin, D.-Y. SARS-CoV-2 NSP13 helicase suppresses interferon signaling by perturbing JAK1 phosphorylation of STAT1. Cell Biosci. 2022, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef]
- Frazier, M.N.; Dillard, L.B.; Krahn, J.M.; Perera, L.; Williams, J.G.; Wilson, I.M.; Stewart, Z.D.; Pillon, M.C.; Deterding, L.J.; Borgnia, M.J.; et al. Characterization of SARS2 Nsp15 nuclease activity reveals it’s mad about U. Nucleic Acids Res. 2021, 49, 10136–10149. [Google Scholar] [CrossRef] [PubMed]
- Vithani, N.; Ward, M.D.; Zimmerman, M.I.; Novak, B.; Borowsky, J.H.; Singh, S.; Bowman, G.R. SARS-CoV-2 Nsp16 activation mechanism and a cryptic pocket with pan-coronavirus antiviral potential. Biophys. J. 2021, 120, 2880–2889. [Google Scholar] [CrossRef] [PubMed]
- Azad, G.K.; Khan, P.K. Variations in Orf3a protein of SARS-CoV-2 alter its structure and function. Biochem. Biophys. Rep. 2021, 26, 100933. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Itoh, Y.; Suzuki, T.; Tanaka, T.; Sakai, Y.; Koido, M.; Hata, C.; Wang, C.-X.; Otani, M.; Moriishi, K.; et al. SARS-CoV-2 ORF6 disrupts nucleocytoplasmic trafficking to advance viral replication. Commun. Biol. 2022, 5, 1–15. [Google Scholar] [CrossRef]
- Redondo, N.; Zaldívar-López, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 2698. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, K.; Qin, B.; Olieric, V.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 Orf9b in complex with human TOM70 suggests unusual virus-host interactions. Nat. Commun. 2021, 12, 2843. [Google Scholar] [CrossRef]
- Suzuki, Y.J.; Gychka, S.G. SARS-CoV-2 Spike Protein Elicits Cell Signaling in Human Host Cells: Implications for Possible Consequences of COVID-19 Vaccines. Vaccines 2021, 9, 36. [Google Scholar] [CrossRef]
- Zhang, Z.; Nomura, N.; Muramoto, Y.; Ekimoto, T.; Uemura, T.; Liu, K.; Yui, M.; Kono, N.; Aoki, J.; Ikeguchi, M.; et al. Structure of SARS-CoV-2 membrane protein essential for virus assembly. Nat. Commun. 2022, 13, 4399. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Cai, Y.; Pang, C.; Wang, L.; McSweeney, S.; Shanklin, J.; Liu, Q. Structural basis for SARS-CoV-2 envelope protein recognition of human cell junction protein PALS1. Nat. Commun. 2021, 12, 3433. [Google Scholar] [CrossRef]
- Mu, J.; Xu, J.; Zhang, L.; Shu, T.; Wu, D.; Huang, M.; Ren, Y.; Li, X.; Geng, Q.; Xu, Y.; et al. SARS-CoV-2-encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci. China Life Sci. 2020, 63, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- RdRp Inhibitors and COVID-19: Is Molnupiravir a Good Option? ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S0753332221013044 (accessed on 19 August 2022).
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.-L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.; Kamil, J.; Lee, B.; Moore, P.; Schulz, T.F.; Muik, A.; Sahin, U.; Türeci, Ö.; Pather, S. The Impact of Evolving SARS-CoV-2 Mutations and Variants on COVID-19 Vaccines. mBio 2022, 13, e0297921. [Google Scholar] [CrossRef] [PubMed]
- Akkiz, H. Implications of the Novel Mutations in the SARS-CoV-2 Genome for Transmission, Disease Severity, and the Vaccine Development. Front. Med. 2021, 8, 636532. [Google Scholar] [CrossRef]
- Lang, Y.; Li, W.; Li, Z.; Koerhuis, D.; van den Burg, A.C.S.; Rozemuller, E.; Bosch, B.-J.; van Kuppeveld, F.J.M.; Boons, G.-J.; Huizinga, E.G.; et al. Coronavirus hemagglutinin-esterase and spike proteins coevolve for functional balance and optimal virion avidity. Proc. Natl. Acad. Sci. USA 2020, 117, 25759–25770. [Google Scholar] [CrossRef]
- Wong, N.A.; Saier, M.H. The SARS-Coronavirus Infection Cycle: A Survey of Viral Membrane Proteins, Their Functional Interactions and Pathogenesis. Int. J. Mol. Sci. 2021, 22, 1308. [Google Scholar] [CrossRef]
- Liu, D.; Tedbury, P.R.; Lan, S.; Huber, A.D.; Puray-Chavez, M.N.; Ji, J.; Michailidis, E.; Saeed, M.; Ndongwe, T.P.; Bassit, L.C.; et al. Visualization of Positive and Negative Sense Viral RNA for Probing the Mechanism of Direct-Acting Antivirals against Hepatitis C Virus. Viruses 2019, 11, 1039. [Google Scholar] [CrossRef]
- Afzal, A. Molecular diagnostic technologies for COVID-19: Limitations and challenges. J. Adv. Res. 2020, 26, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2020, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Nagesha, S.N.; Ramesh, B.N.; Pradeep, C.; Shashidhara, K.S.; Ramakrishnappa, T.; Krishnaprasad, B.T.; Jnanashree, S.M.; Manohar, M.; Arunkumar, N.; Patel, D.; et al. SARS-CoV 2 spike protein S1 subunit as an ideal target for stable vaccines: A bioinformatic study. Mater. Today Proc. 2022, 49, 904–912. [Google Scholar] [CrossRef]
- Thomas, S. The Structure of the Membrane Protein of SARS-CoV-2 Resembles the Sugar Transporter SemiSWEET. Pathog. Immun. 2020, 5, 342–363. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Liu, G.; Ma, H.; Zhao, D.; Yang, Y.; Liu, M.; Mohammed, A.; Zhao, C.; Yang, Y.; Xie, J.; et al. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem. Biophys. Res. Commun. 2020, 527, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef]
- Fitzgerald, D.M.; Rosenberg, S.M. What is mutation? A chapter in the series: How microbes “jeopardize” the modern synthesis. PLoS Genet. 2019, 15, e1007995. [Google Scholar] [CrossRef]
- Mallapaty, S. Did the coronavirus jump from animals to people twice? Nature 2021, 597, 458–459. [Google Scholar] [CrossRef]
- Halley, J.M.; Vokou, D.; Pappas, G.; Sainis, I. SARS-CoV-2 mutational cascades and the risk of hyper-exponential growth. Microb. Pathog. 2021, 161, 105237. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.N.; Fearns, R. Genetic Instability of RNA Viruses. Genome Stab. 2016, 21–35. [Google Scholar] [CrossRef]
- Islam, A.; Shahi, S.; Marzan, A.A.; Amin, M.R.; Hasan, M.N.; Hoque, M.N.; Ghosh, A.; Barua, A.; Khan, A.; Dhama, K.; et al. Variant-specific deleterious mutations in the SARS-CoV-2 genome reveal immune responses and potentials for prophylactic vaccine development. Front. Pharmacol. 2023, 14, 1090717. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Eguia, R.T.; Crawford, K.H.D.; Stevens-Ayers, T.; Kelnhofer-Millevolte, L.; Greninger, A.L.; Englund, J.A.; Boeckh, M.J.; Bloom, J.D. A human coronavirus evolves antigenically to escape antibody immunity. PLoS Pathog. 2021, 17, e1009453. [Google Scholar] [CrossRef] [PubMed]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, I.; Pravica, V.; Miljanovic, D.; Cupic, M. Immune Evasion of SARS-CoV-2 Emerging Variants: What Have We Learnt So Far? Viruses 2021, 13, 1192. [Google Scholar] [CrossRef]
- Mohammadi, E.; Shafiee, F.; Shahzamani, K.; Ranjbar, M.M.; Alibakhshi, A.; Ahangarzadeh, S.; Beikmohammadi, L.; Shariati, L.; Hooshmandi, S.; Ataei, B.; et al. Novel and emerging mutations of SARS-CoV-2: Biomedical implications. Biomed. Pharmacother. 2021, 139, 111599. [Google Scholar] [CrossRef]
- Manathunga, S.S.; Abeyagunawardena, I.A.; Dharmaratne, S.D. A comparison of transmissibility of SARS-CoV-2 variants of concern. Virol. J. 2023, 20, 59. [Google Scholar] [CrossRef]
- Padhan, K.; Parvez, M.K.; Al-Dosari, M.S. Comparative sequence analysis of SARS-CoV-2 suggests its high transmissibility and pathogenicity. Future Virol. 2021, 16, 245–254. [Google Scholar] [CrossRef]
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D.; et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct. Target. Ther. 2022, 7, 138. [Google Scholar] [CrossRef] [PubMed]
- LaPelusa, A.; Kaushik, R. Physiology, Proteins. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK555990/ (accessed on 26 October 2022).
- Zhang, Z.; Liu, W.; Zhang, S.; Wei, P.; Zhang, L.; Chen, D.; Qiu, S.; Li, X.; Zhao, J.; Shi, Y.; et al. Two novel human coronavirus OC43 genotypes circulating in hospitalized children with pneumonia in China. Emerg. Microbes Infect. 2021, 11, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hesketh, E.L.; Shamorkina, T.M.; Li, W.; Franken, P.J.; Drabek, D.; van Haperen, R.; Townend, S.; van Kuppeveld, F.J.M.; Grosveld, F.; et al. Antigenic structure of the human coronavirus OC43 spike reveals exposed and occluded neutralizing epitopes. Nat. Commun. 2022, 13, 2921. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Jaimes, J.A.; Whittaker, G.R. Molecular diversity of coronavirus host cell entry receptors. FEMS Microbiol. Rev. 2020, 45, fuaa057. [Google Scholar] [CrossRef]
- Nassar, A.; Ibrahim, I.M.; Amin, F.G.; Magdy, M.; Elgharib, A.M.; Azzam, E.B.; Nasser, F.; Yousry, K.; Shamkh, I.M.; Mahdy, S.M.; et al. A Review of Human Coronaviruses’ Receptors: The Host-Cell Targets for the Crown Bearing Viruses. Molecules 2021, 26, 6455. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Jemimah, S.; Ponnuswamy, P.K.; Gromiha, M.M. Why are ACE2 binding coronavirus strains SARS-CoV/SARS-CoV-2 wild and NL63 mild? Proteins Struct. Funct. Bioinform. 2021, 89, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Samavati, L.; Uhal, B.D. ACE2, Much More Than Just a Receptor for SARS-CoV-2. Front. Cell. Infect. Microbiol. 2020, 10, 554397. [Google Scholar] [CrossRef]
- González-Morelo, K.J.; Vega-Sagardía, M.; Garrido, D. Molecular Insights Into O-Linked Glycan Utilization by Gut Microbes. Front. Microbiol. 2020, 11, 591568. [Google Scholar] [CrossRef]
- Hulswit, R.J.G.; Lang, Y.; Bakkers, M.J.G.; Li, W.; Li, Z.; Schouten, A.; Ophorst, B.; van Kuppeveld, F.J.M.; Boons, G.-J.; Bosch, B.-J.; et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc. Natl. Acad. Sci. USA 2019, 116, 2681–2690. [Google Scholar] [CrossRef]
- Lachance, C.; Arbour, N.; Cashman, N.R.; Talbot, P.J. Involvement of Aminopeptidase N (CD13) in Infection of Human Neural Cells by Human Coronavirus 229E. J. Virol. 1998, 72, 6511–6519. [Google Scholar] [CrossRef]
- Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Prokscha, A.; Naim, H.Y.; Müller, M.A.; Drosten, C.; Pöhlmann, S.; Hoffmann, M. Polymorphisms in dipeptidyl peptidase 4 reduce host cell entry of Middle East respiratory syndrome coronavirus. Emerg. Microbes Infect. 2020, 9, 155–168. [Google Scholar] [CrossRef] [PubMed]
- de Castro, K.C.; Costa, J.M. Polymeric surfaces with biocidal action: Challenges imposed by the SARS-CoV-2, technologies employed, and future perspectives. J. Polym. Res. 2021, 28, 230. [Google Scholar] [CrossRef]
- Paintsil, E.; Cheng, Y.-C. Antiviral Agents. In Encyclopedia of Microbiology; Elsevier: Amsterdam, The Netherlands, 2009; pp. 223–257. [Google Scholar] [CrossRef]
- Pruijssers, A.J.; Denison, M.R. Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 2019, 35, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Jing, X.; Zhang, P.; De Clercq, E. Antiviral Classification. Encycl. Virol. 2021, 121–130. [Google Scholar] [CrossRef]
- Lenard, J. Viral Membranes. Encycl. Virol. 2008, 308–314. [Google Scholar] [CrossRef]
- Nucleoside Analogues. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012. Available online: http://www.ncbi.nlm.nih.gov/books/NBK548938/ (accessed on 30 September 2022).
- Weber, I.T.; Kneller, D.W.; Wong-Sam, A. Highly resistant HIV-1 proteases and strategies for their inhibition. Future Med. Chem. 2015, 7, 1023–1038. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Sheida, A.; Taghizadieh, M.; Memar, M.Y.; Hamblin, M.R.; Bannazadeh Baghi, H.; Sadri Nahand, J.; Asemi, Z.; Mirzaei, H. Paxlovid (Nirmatrelvir/Ritonavir): A new approach to COVID-19 therapy? Biomed. Pharmacother. 2023, 162, 114367. [Google Scholar] [CrossRef]
- Midde, N.M.; Patters, B.J.; Rao, P.; Cory, T.J.; Kumar, S. Investigational protease inhibitors as antiretroviral therapies. Expert. Opin. Investig. Drugs 2016, 25, 1189–1200. [Google Scholar] [CrossRef][Green Version]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS (Auckl) 2015, 7, 95–104. [Google Scholar] [CrossRef]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef]
- Zenchenko, A.A.; Drenichev, M.S.; Il’icheva, I.A.; Mikhailov, S.N. Antiviral and Antimicrobial Nucleoside Derivatives: Structural Features and Mechanisms of Action. Mol. Biol. 2021, 55, 786–812. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, K.; Daikoku, T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol. Ther. 2020, 209, 107512. [Google Scholar] [CrossRef] [PubMed]
- Nirwan, S.; Kakkar, R. Rhinovirus RNA Polymerase. Viral Polym. 2019, 301–331. [Google Scholar] [CrossRef]
- Padhi, A.K.; Dandapat, J.; Saudagar, P.; Uversky, V.N.; Tripathi, T. Interface-based design of the favipiravir-binding site in SARS-CoV-2 RNA-dependent RNA polymerase reveals mutations conferring resistance to chain termination. FEBS Lett. 2021, 595, 2366–2382. [Google Scholar] [CrossRef] [PubMed]
- PubChem Favipiravir. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/492405 (accessed on 2 December 2023).
- ChemDraw—PerkinElmer Informatics. Available online: https://perkinelmerinformatics.com/products/research/chemdraw (accessed on 15 May 2023).
- Sirijatuphat, R.; Manosuthi, W.; Niyomnaitham, S.; Owen, A.; Copeland, K.K.; Charoenpong, L.; Rattanasompattikul, M.; Mahasirimongkol, S.; Wichukchinda, N.; Chokephaibulkit, K. Early treatment of Favipiravir in COVID-19 patients without pneumonia: A multicentre, open-labelled, randomized control study. Emerg. Microbes Infect. 2022, 11, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.L.; Orton, C.M.; Grinsztejn, B.; Donaldson, G.C.; Ramírez, B.C.; Tonkin, J.; Santos, B.R.; Cardoso, S.W.; Ritchie, A.I.; Conway, F.; et al. Favipiravir in patients hospitalised with COVID-19 (PIONEER trial): A multicentre, open-label, phase 3, randomised controlled trial of early intervention versus standard care. Lancet Respir. Med. 2023, 11, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Sluis-Cremer, N. Future of nonnucleoside reverse transcriptase inhibitors. Proc. Natl. Acad. Sci. USA 2018, 115, 637–638. [Google Scholar] [CrossRef]
- Rehman, N.; Nguyen, H. Nevirapine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK554477/ (accessed on 11 October 2022).
- Rock, B.M.; Hengel, S.M.; Rock, D.A.; Wienkers, L.C.; Kunze, K.L. Characterization of ritonavir-mediated inactivation of cytochrome P450 3A4. Mol. Pharmacol. 2014, 86, 665–674. [Google Scholar] [CrossRef]
- PubChem PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 11 October 2022).
- Heidary, M.; Asadi, A.; Noorbakhsh, N.; Dashtbin, S.; Asadollahi, P.; Dranbandi, A.; Navidifar, T.; Ghanavati, R. COVID-19 in HIV-positive patients: A systematic review of case reports and case series. J. Clin. Lab. Anal. 2022, 36, e24308. [Google Scholar] [CrossRef]
- Identification of Pyrogallol as a Warhead in Design of Covalent Inhibitors for the SARS-CoV-2 3CL Protease|Nature Communications. Available online: https://www.nature.com/articles/s41467-021-23751-3 (accessed on 27 October 2022).
- Duan, X.; Lacko, L.A.; Chen, S. Druggable targets and therapeutic development for COVID-19. Front. Chem. 2022, 10, 963701. [Google Scholar] [CrossRef]
- Lv, Z.; Cano, K.E.; Jia, L.; Drag, M.; Huang, T.T.; Olsen, S.K. Targeting SARS-CoV-2 Proteases for COVID-19 Antiviral Development. Front. Chem. 2022, 9, 819165. [Google Scholar] [CrossRef] [PubMed]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti–COVID-19 drug design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef] [PubMed]
- Schuller, M.; Zarganes-Tzitzikas, T.; Bennett, J.; De Cesco, S.; Fearon, D.; von Delft, F.; Fedorov, O.; Brennan, P.E.; Ahel, I. Discovery and Development Strategies for SARS-CoV-2 NSP3 Macrodomain Inhibitors. Pathogens 2023, 12, 324. [Google Scholar] [CrossRef] [PubMed]
- Roe, M.K.; Junod, N.A.; Young, A.R.; Beachboard, D.C.; Stobart, C.C. Targeting novel structural and functional features of coronavirus protease nsp5 (3CLpro, Mpro) in the age of COVID-19. J. Gen. Virol. 2021, 102, 001558. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef]
- Liu, J.; Pan, X.; Zhang, S.; Li, M.; Ma, K.; Fan, C.; Lv, Y.; Guan, X.; Yang, Y.; Ye, X.; et al. Efficacy and safety of Paxlovid in severe adult patients with SARS-Cov-2 infection: A multicenter randomized controlled study. Lancet Reg. Health West. Pac. 2023, 33, 100694. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef]
- PubChem Nirmatrelvir. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/155903259 (accessed on 28 December 2023).
- Amani, B.; Amani, B. Efficacy and safety of nirmatrelvir/ritonavir (Paxlovid) for COVID-19: A rapid review and meta-analysis. J. Med. Virol. 2023, 95, e28441. [Google Scholar] [CrossRef]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef]
- Kokic, G.; Hillen, H.S.; Tegunov, D.; Dienemann, C.; Seitz, F.; Schmitzova, J.; Farnung, L.; Siewert, A.; Höbartner, C.; Cramer, P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H. Viral Polymerases. Adv. Exp. Med. Biol. 2012, 726, 267–304. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Götte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef] [PubMed]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Chen, C.; Tang, J.; Wang, C.; Zhou, M.; Cheng, Y.; Zhou, X.; Wu, Q.; Zhang, X.; Feng, Z.; et al. Efficacy and safety of three new oral antiviral treatment (molnupiravir, fluvoxamine and Paxlovid) for COVID-19: A meta-analysis. Ann. Med. 2022, 54, 516–523. [Google Scholar] [CrossRef]
- Zarenezhad, E.; Marzi, M. Review on molnupiravir as a promising oral drug for the treatment of COVID-19. Med. Chem. Res. 2022, 31, 232–243. [Google Scholar] [CrossRef]
- Venkataraman, S.; Prasad, B.V.L.S.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef]
- PubChem Remdesivir. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/121304016 (accessed on 28 December 2023).
- PubChem Molnupiravir. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/145996610 (accessed on 28 December 2023).
- Gupte, V.; Hegde, R.; Sawant, S.; Kalathingal, K.; Jadhav, S.; Malabade, R.; Gogtay, J. Safety and clinical outcomes of remdesivir in hospitalised COVID-19 patients: A retrospective analysis of active surveillance database. BMC Infect. Dis. 2022, 22, 1. [Google Scholar] [CrossRef]
- Clinical Antiviral Efficacy of Remdesivir and Casirivimab/Imdevimab against the SARS-CoV-2 Delta and Omicron Variants|medRxiv. Available online: https://www.medrxiv.org/content/10.1101/2022.10.17.22281161v1.full-text (accessed on 1 September 2023).
- Challenges and Opportunities for Antiviral Monoclonal Antibodies as COVID-19 Therapy—ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S0169409X20302751 (accessed on 27 October 2022).
- Aleem, A.; Slenker, A.K. Monoclonal Antibody Therapy For High-Risk Coronavirus (COVID 19) Patients with Mild To Moderate Disease Presentations. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK570603/ (accessed on 30 March 2022).
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef]
- Ramesh, D.; Vijayakumar, B.G.; Kannan, T. Advances in Nucleoside and Nucleotide Analogues in Tackling Human Immunodeficiency Virus and Hepatitis Virus Infections. ChemMedChem 20212, 16, 1403–1419. [Google Scholar] [CrossRef]
- Melo-Filho, C.C.; Bobrowski, T.; Martin, H.-J.; Sessions, Z.; Popov, K.I.; Moorman, N.J.; Baric, R.S.; Muratov, E.N.; Tropsha, A. Conserved coronavirus proteins as targets of broad-spectrum antivirals. Antivir. Res. 2022, 204, 105360. [Google Scholar] [CrossRef]
- Punekar, M.; Kshirsagar, M.; Tellapragada, C.; Patil, K. Repurposing of antiviral drugs for COVID-19 and impact of repurposed drugs on the nervous system. Microb. Pathog. 2022, 168, 105608. [Google Scholar] [CrossRef]
- Heo, Y.-A. Sotrovimab: First Approval. Drugs 2022, 82, 477–484. [Google Scholar] [CrossRef] [PubMed]
- FDA. Coronavirus (COVID-19)|Drugs. 2023. Available online: https://www.fda.gov/drugs/emergency-preparedness-drugs/coronavirus-covid-19-drugs (accessed on 30 August 2023).
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171–1187.e20. [Google Scholar] [CrossRef] [PubMed]
- Lasheras, I.; Santabárbara, J. Use of antimalarial drugs in the treatment of COVID-19: A window of opportunity? Med. Clin. (Engl. Ed.) 2020, 155, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Shibeshi, M.A.; Kifle, Z.D.; Atnafie, S.A. Antimalarial Drug Resistance and Novel Targets for Antimalarial Drug Discovery. Infect. Drug Resist. 2020, 13, 4047–4060. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.-C.; Wang, Y.-H.; Chen, Y.-L.; Tsai, W.-C.; Lee, C.-H.; Chuang, K.-P.; Chen, Y.-M.A.; Yuan, C.-H.; Ho, S.-Y.; Yang, M.-H.; et al. Chloroquine and Hydroxychloroquine: Efficacy in the Treatment of the COVID-19. Pathogens 2021, 10, 217. [Google Scholar] [CrossRef]
- Axfors, C.; Schmitt, A.M.; Janiaud, P.; van’t Hooft, J.; Abd-Elsalam, S.; Abdo, E.F.; Abella, B.S.; Akram, J.; Amaravadi, R.K.; Angus, D.C.; et al. Mortality outcomes with hydroxychloroquine and chloroquine in COVID-19 from an international collaborative meta-analysis of randomized trials. Nat. Commun. 2021, 12, 2349. [Google Scholar] [CrossRef]
- Kausar, S.; Said Khan, F.; Ishaq Mujeeb Ur Rehman, M.; Akram, M.; Riaz, M.; Rasool, G.; Hamid Khan, A.; Saleem, I.; Shamim, S.; Malik, A. A review: Mechanism of action of antiviral drugs. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211002620. [Google Scholar] [CrossRef]
- CDC. Centers for Disease Control and Prevention. 2020; COVID Data Tracker. Available online: https://covid.cdc.gov/covid-data-tracker (accessed on 24 October 2022).
- Johnson, A.G. COVID-19 Incidence and Death Rates Among Unvaccinated and Fully Vaccinated Adults with and without Booster Doses During Periods of Delta and Omicron Variant Emergence—25 U.S. Jurisdictions, April 4–December 25, 2021. MMWR. Morb. Mortal. Wkly. Rep. 2022, 71, 132–138. Available online: https://www.cdc.gov/mmwr/volumes/71/wr/mm7104e2.htm (accessed on 24 October 2022). [CrossRef]
- Malik, J.A.; Ahmed, S.; Mir, A.; Shinde, M.; Bender, O.; Alshammari, F.; Ansari, M.; Anwar, S. The SARS-CoV-2 mutations versus vaccine effectiveness: New opportunities to new challenges. J. Infect. Public. Health 2022, 15, 228–240. [Google Scholar] [CrossRef]
- Abbasian, M.H.; Mahmanzar, M.; Rahimian, K.; Mahdavi, B.; Tokhanbigli, S.; Moradi, B.; Sisakht, M.M.; Deng, Y. Global landscape of SARS-CoV-2 mutations and conserved regions. J. Transl. Med. 2023, 21, 152. [Google Scholar] [CrossRef] [PubMed]
- Jaroszewski, L.; Iyer, M.; Alisoltani, A.; Sedova, M.; Godzik, A. The interplay of SARS-CoV-2 evolution and constraints imposed by the structure and functionality of its proteins. PLoS Comput. Biol. 2021, 17, e1009147. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Naiyer, S.; Mansuri, S.; Soni, N.; Singh, V.; Bhat, K.H.; Singh, N.; Arora, G.; Mansuri, M.S. COVID-19 Diagnosis: A Comprehensive Review of the RT-qPCR Method for Detection of SARS-CoV-2. Diagnostics 2022, 12, 1503. [Google Scholar] [CrossRef] [PubMed]
- Amanat, F.; White, K.M.; Miorin, L.; Strohmeier, S.; McMahon, M.; Meade, P.; Liu, W.; Albrecht, R.A.; Simon, V.; Martinez-Sobrido, L.; et al. An In Vitro Microneutralization Assay for SARS-CoV-2 Serology and Drug Screening. Curr. Protoc. Microbiol. 2020, 58, e108. [Google Scholar] [CrossRef]
- Bhatia, R. Addressing challenge of zoonotic diseases through One Health approach. Indian. J. Med. Res. 2021, 153, 249–252. [Google Scholar] [CrossRef]
- Najjar-Debbiny, R.; Gronich, N.; Weber, G.; Khoury, J.; Amar, M.; Stein, N.; Goldstein, L.H.; Saliba, W. Effectiveness of Paxlovid in Reducing Severe Coronavirus Disease 2019 and Mortality in High-Risk Patients. Clin. Infect. Dis. 2023, 76, e342–e349. [Google Scholar] [CrossRef]
- Kudlay, D.; Svistunov, A. COVID-19 Vaccines: An Overview of Different Platforms. Bioengineering 2022, 9, 72. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Outteridge, M.; Nunn, C.M.; Devine, K.; Patel, B.; McLean, G.R. Antivirals for Broader Coverage against Human Coronaviruses. Viruses 2024, 16, 156. https://doi.org/10.3390/v16010156
Outteridge M, Nunn CM, Devine K, Patel B, McLean GR. Antivirals for Broader Coverage against Human Coronaviruses. Viruses. 2024; 16(1):156. https://doi.org/10.3390/v16010156
Chicago/Turabian StyleOutteridge, Mia, Christine M. Nunn, Kevin Devine, Bhaven Patel, and Gary R. McLean. 2024. "Antivirals for Broader Coverage against Human Coronaviruses" Viruses 16, no. 1: 156. https://doi.org/10.3390/v16010156
APA StyleOutteridge, M., Nunn, C. M., Devine, K., Patel, B., & McLean, G. R. (2024). Antivirals for Broader Coverage against Human Coronaviruses. Viruses, 16(1), 156. https://doi.org/10.3390/v16010156