
The Identification of Viral Pathogens in a Physostegia virginiana Plant Using High-Throughput RNA Sequencing




Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Samples and Plant Growth
2.2. RNA Sequencing (RNA-Seq) and De Novo Assembly
2.3. PCR, RT-PCR, and Rapid Amplification of cDNA Ends (RACE)
2.4. Analyses of Viral Genome and Proteins
2.5. Phylogenetic Analysis
2.6. Mechanical Inoculation
3. Results
3.1. Identification of Five Viruses in Physostegia virginiana through RNA-Seq
3.2. Determination of Genomic Sequences of the Four New Viruses
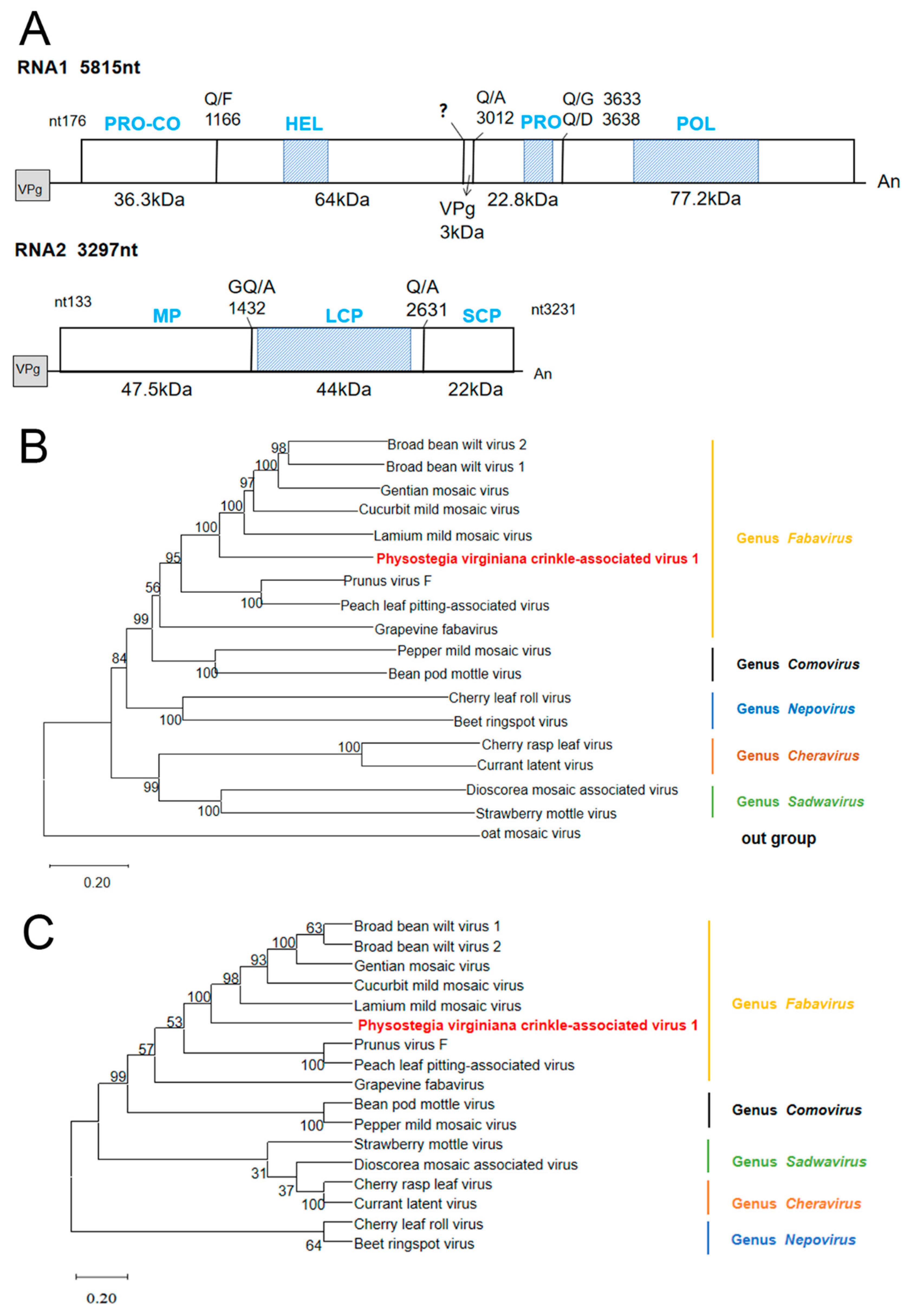
3.3. Genomic Structure and Phylogenetic Analysis of PVCaV1
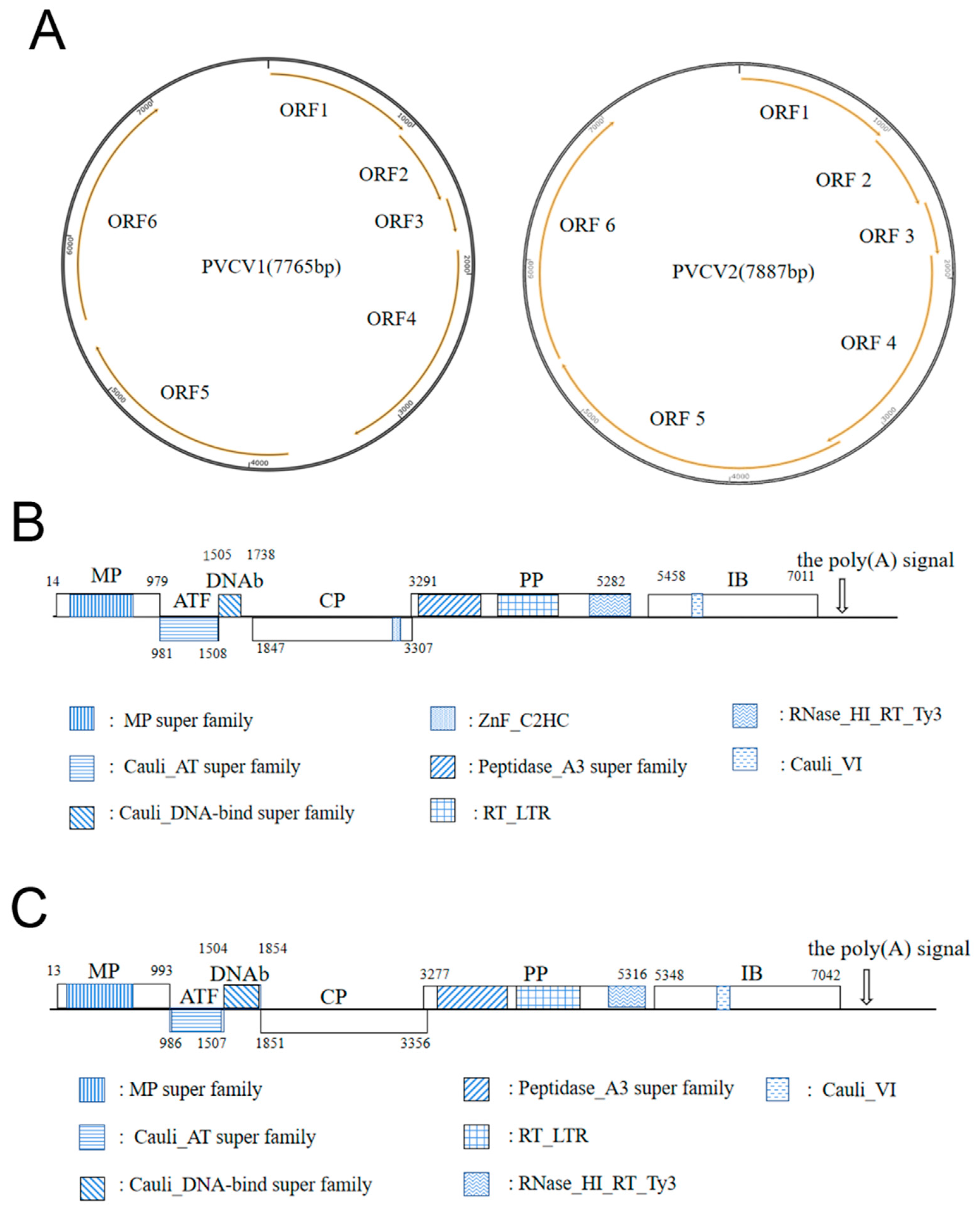
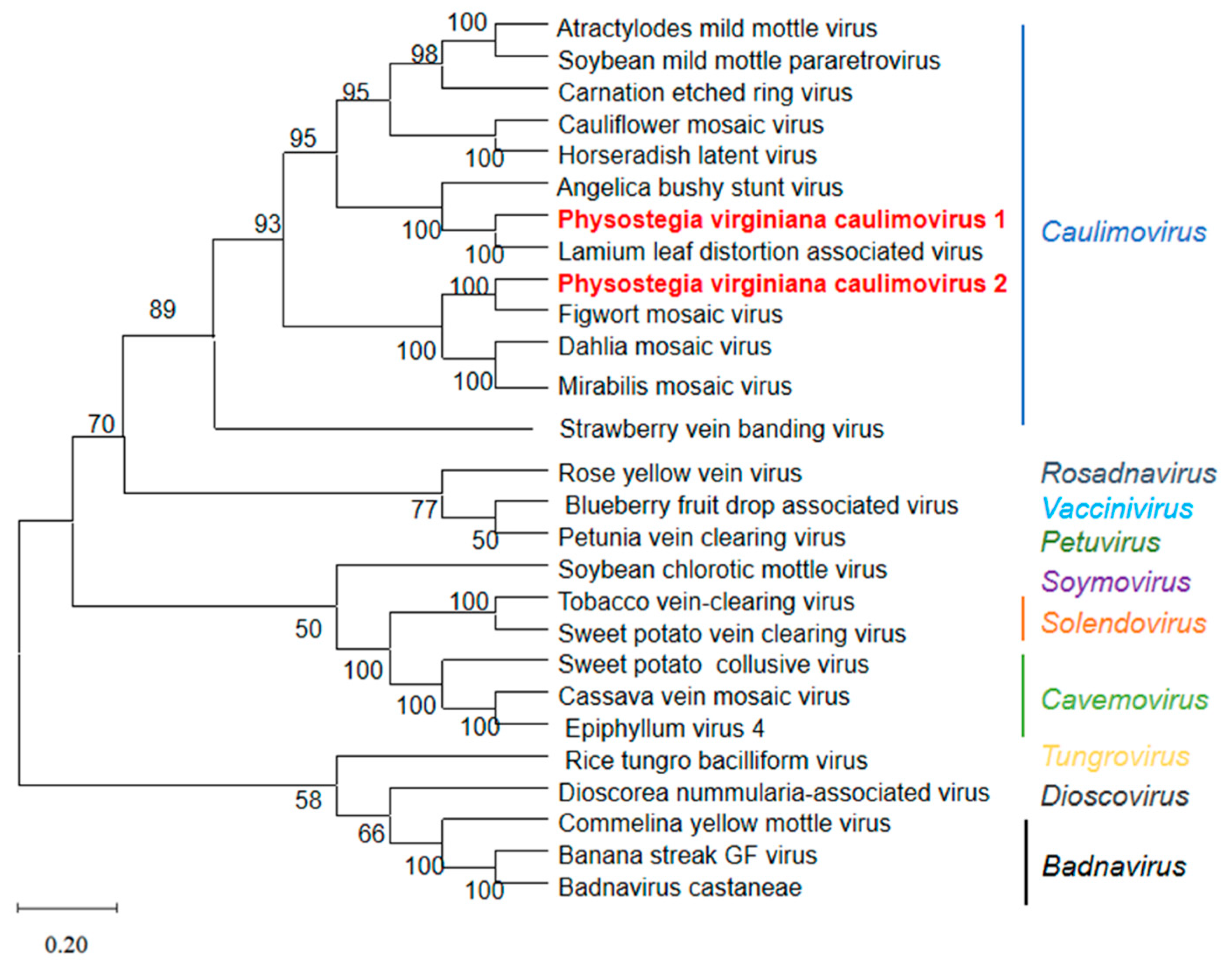
3.4. Genomic Structure and Phylogenetic Analyses of PVCV1 and PVCV2
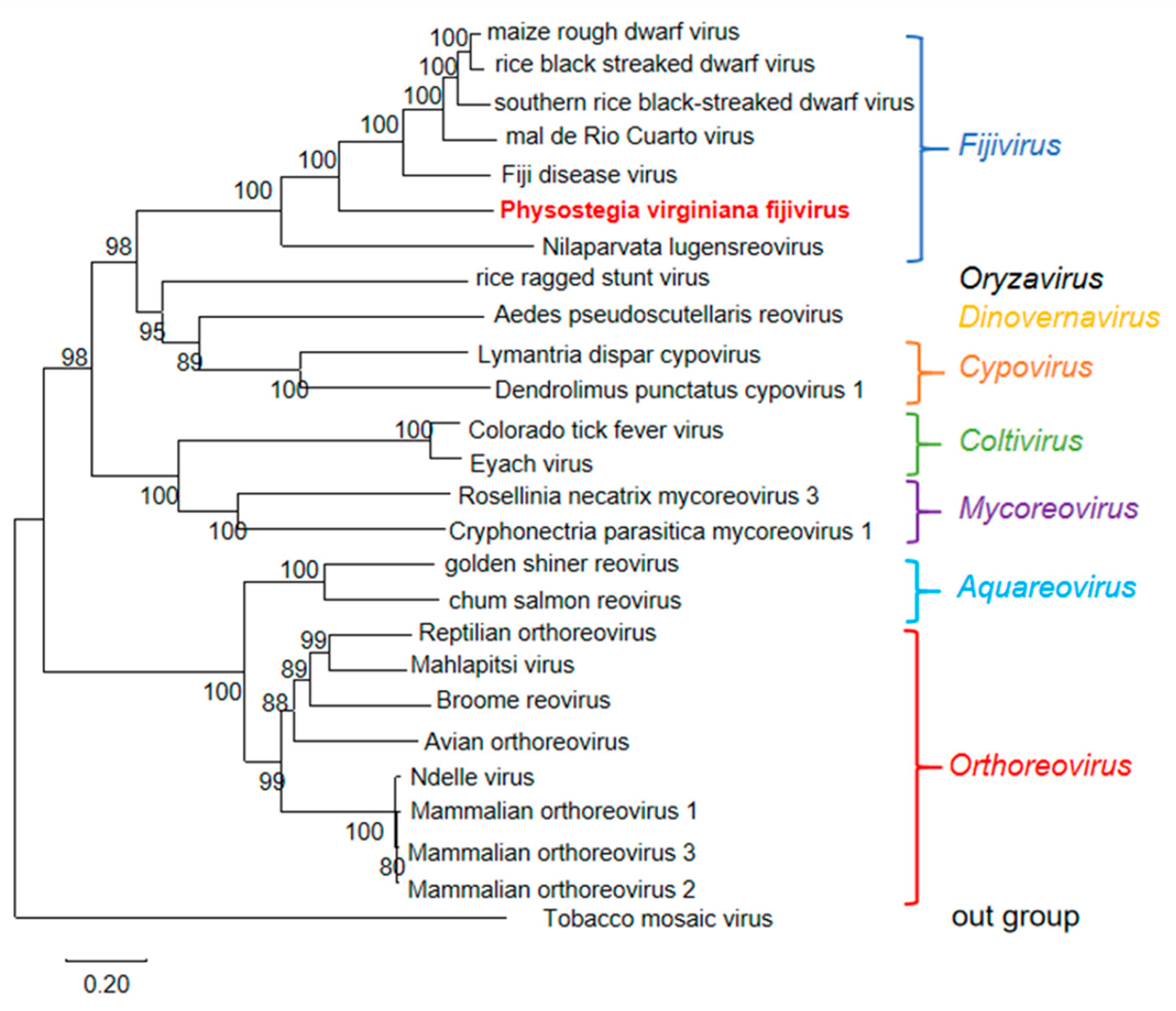
3.5. Genomic Structure and Phylogenetic Analyses of PVFV
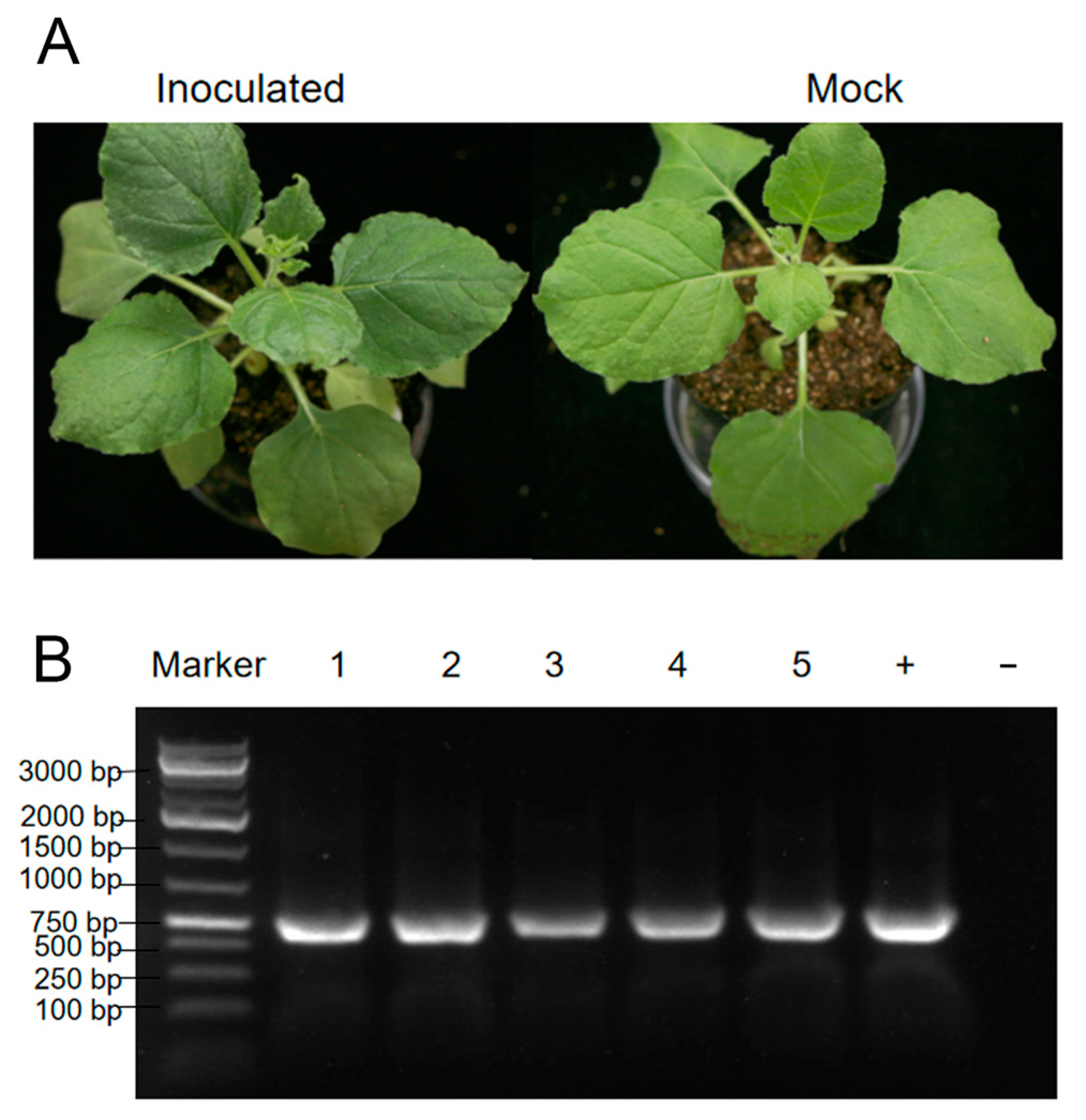
3.6. Mechanical Inoculation of PVCaV1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, H.Z.; Jiang, F.S.; Lan, Y.B.; Han, X. First Record of Paramyrothecium roridum Causing Leaf Spot on Physostegia virginiana in China. Plant Dis. 2021, 105, 505. [Google Scholar] [CrossRef]
- Cardin, L.; Onesto, J.P.; Moury, B. First Report of Alfalfa mosaic virus in Physostegia virginiana. Plant Dis. 2002, 86, 72. [Google Scholar] [CrossRef]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef]
- Engvall, E.; Perlmann, P. Enzyme-linked immunosorbent assay (ELISA) quantitative assay of immunoglobulin G. Immunochemistry 1971, 8, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Hadidi, A.; Flores, R.; Candresse, T.; Barba, M. Next-Generation Sequencing and Genome Editing in Plant Virology. Front. Microbiol. 2016, 7, 1325. [Google Scholar] [CrossRef] [PubMed]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rott, M.; Xiang, Y.; Boyes, I.; Belton, M.; Saeed, H.; Kesanakurti, P.; Hayes, S.; Lawrence, T.; Birch, C.; Bhagwat, B.; et al. Application of Next Generation Sequencing for Diagnostic Testing of Tree Fruit Viruses and Viroids. Plant Dis. 2017, 101, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fu, S.; Wu, H.Y.; Cao, M.J.; Liu, L.; Zhou, X.P.; Wu, J.X. Discovery and genomic function of a novel rice dwarf-associated bunya-like virus. Viruses 2022, 14, 1183. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Fu, S.; Xie, L.; Wang, Y.Q.; Cao, M.J.; Zhou, X.P.; Wu, J.X. Identification and characterization of two novel noda-like viruses from rice plants showing the dwarfing symptom. Viruses 2022, 14, 1159. [Google Scholar] [CrossRef]
- Chu, B.; Anane, R.F.; Li, S.; Gao, L.; Zi, S.; Yan, K.; Ji, K.; Chen, Z.; Zhao, M. Complete genome sequence analysis of Valeriana jatamansi tymovirus 1: A novel member of the genus Tymovirus infecting Valeriana jatamansi Jones. Arch. Virol. 2023, 168, 245. [Google Scholar] [CrossRef]
- Cao, X.; Wang, Z.; Pang, J.; Sun, L.; Kondo, H.; Andika, I.B. Identification of a novel dicistro-like virus associated with the roots of tomato plants. Arch. Virol. 2023, 168, 214. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Hily, J.-M.; Petrzik, K.; Sanfaçon, H.; Thompson, J.R.; van der Vlugt, R.; Wetzel, T.; Consortium, I.R. ICTV Virus Taxonomy Profile: Secoviridae 2022. J. Gen. Virol. 2022, 103, 001807. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, R.M.; Guerri, J.; Luis-Arteaga, M.S.; Moreno, P.; Rubio, L. The complete sequence of a Spanish isolate of Broad bean wilt virus 1 (BBWV-1) reveals a high variability and conserved motifs in the genus Fabavirus. Arch. Virol. 2005, 150, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Carrier, K.; Hans, F.; Sanfaçon, H. Mutagenesis of Amino Acids at Two Tomato Ringspot Nepovirus Cleavage Sites: Effect on Proteolytic Processing in cis and in trans by the 3C-like Protease. Virology 1999, 258, 161–175. [Google Scholar] [CrossRef]
- Ferriol, I.; Silva Junior, D.M.; Nigg, J.C.; Zamora-Macorra, E.J.; Falk, B.W. Identification of the cleavage sites of the RNA2-encoded polyproteins for two members of the genus Torradovirus by N-terminal sequencing of the virion capsid proteins. Virology 2016, 498, 109–115. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Donchenko, A.P.; Blinov, V.M.; Koonin, E.V. Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases. FEBS Lett. 1989, 243, 103–114. [Google Scholar] [CrossRef]
- Margis, R.; Pinck, L. Effects of site-directed mutagenesis on the presumed catalytic triad and substrate-binding pocket of grapevine fanleaf nepovirus 24-kDa proteinase. Virology 1992, 190, 884–888. [Google Scholar] [CrossRef]
- Thole, V.; Hull, R. Rice Tungro Spherical Virus Polyprotein Processing: Identification of a Virus-Encoded Protease and Mutational Analysis of Putative Cleavage Sites. Virology 1998, 247, 106–114. [Google Scholar] [CrossRef]
- Wellink, J.; van Kammen, A. Proteases involved in the processing of viral polyproteins. Arch. Virol. 1988, 98, 1–26. [Google Scholar] [CrossRef]
- Igori, D.; Shin, A.-Y.; Hwang, U.S.; Choi, E.K.; Kwon, S.-Y.; Moon, J.S. Genome sequence and characterization of a novel fabavirus infecting Cirsium setidens (gondre) in South Korea. Arch. Virol. 2023, 168, 77. [Google Scholar] [CrossRef]
- Koh, L.H.; Cooper, J.I.; Wong, S.M. Complete sequences and phylogenetic analyses of a Singapore isolate of broad bean wilt fabavirus. Arch. Virol. 2001, 146, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Pooggin, M.M.; Fütterer, J.; Skryabin, K.G.; Hohn, T. A short open reading frame terminating in front of a stable hairpin is the conserved feature in pregenomic RNA leaders of plant pararetroviruses. J. Gen. Virol. 1999, 80, 2217–2228. [Google Scholar] [CrossRef] [PubMed]
- Pappu, H.R.; Hammett, K.R.W.; Druffel, K.L. Dahlia mosaic virus and Tobacco streak virus in Dahlia (Dahlia variabilis) in New Zealand. Plant Dis. 2008, 92, 1138. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Fang, S.G.; Xu, J.L.; Sun, L.Y.; Li, D.W.; Yu, J.L. Sequence Analysis of the Complete Genome of Rice Black-Streaked Dwarf Virus Isolated from Maize with Rough Dwarf Disease. Virus Genes 2003, 27, 163–168. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2014, 43, D222–D226. [Google Scholar] [CrossRef]
- Kobayashi, Y.O.; Kobayashi, A.; Nakano, M.; Hagiwara, K.; Honda, Y.; Omura, T. Analysis of genetic relations between Broad bean wilt virus 1 and Broad bean wilt virus 2. J. Gen. Plant Pathol. 2003, 69, 320–326. [Google Scholar] [CrossRef]
- Dong, S.W.; Xiang, H.Y.; Shang, Q.X.; Li, D.W.; Yu, J.L.; Han, C.G. Complete genomic sequence analysis reveals a novel fabavirus infecting cucurbits in China. Arch. Virol. 2012, 157, 597–600. [Google Scholar] [CrossRef]
- Qi, Y.J.; Zhou, X.P.; Xue, C.Y.; Li, D.B. Nucleotide sequence of RNA2 and polyprotein processing sites of a Chinese isolate of broad bean wilt virus 2. Prog. Nat. Sci. 2000, 10, 680–686. [Google Scholar]
- Villamor, D.E.V.; Pillai, S.S.; Eastwell, K.C. High throughput sequencing reveals a novel fabavirus infecting sweet cherry. Arch. Virol. 2017, 162, 811–816. [Google Scholar] [CrossRef]
- Yoo, R.H.; Zhao, F.; Lim, S.; Igori, D.; Lee, S.H.; Moon, J.S. The complete nucleotide sequence and genome organization of lychnis mottle virus. Arch. Virol. 2015, 160, 2891–2894. [Google Scholar] [CrossRef]
- Zhou, X.P.; Qi, Y.J.; Li, F.; Huang, X.Z.; Li, D.B. Complete Nucleotide Sequence and Possible Genomic Expression Strategy of a Chinese Isolate of Broad Bean Wilt Virus. Acta Biochim. Et Biophys. Sin. 2001, 33, 46–52. [Google Scholar]
- Liu, C.K.; Ye, L.F.; Lang, G.J.; Zhang, C.X.; Hong, J.; Zhou, X.P. The VP37 protein of Broad bean wilt virus 2 induces tubule-like structures in both plant and insect cells. Virus Res. 2011, 155, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.S.; Chisholm, J.; Sanfaçon, H. Strawberry mottle virus (family Secoviridae, order Picornavirales) encodes a novel glutamic protease to process the RNA2 polyprotein at two cleavage sites. J. Virol. 2019, 93, e01679-18. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.X.; Sun, J.; Zhang, S.; Wang, J.Z.; Zhang, S.Y.; Yu, M.C.; Cao, M.J. Complete genome sequence of a putative new and distinct caulimovirus from Metaplexis japonica (Thunb.) Makino in China. Arch. Virol. 2021, 166, 3433–3436. [Google Scholar] [CrossRef]
- Teycheney, P.-Y.; Geering, A.D.W.; Dasgupta, I.; Hull, R.; Kreuze, J.F.; Lockhart, B.; Muller, E.; Olszewski, N.; Pappu, H.; Pooggin, M.M.; et al. ICTV Virus Taxonomy Profile: Caulimoviridae. J. Gen. Virol. 2020, 101, 1025–1026. [Google Scholar] [CrossRef]
- Lim, S.; Baek, D.; Igori, D.; Moon, J.S. Complete genome sequence of a putative new caulimovirus which exists as endogenous pararetroviral sequences in Angelica dahurica. Arch. Virol. 2017, 162, 3837–3842. [Google Scholar] [CrossRef]
- Pappu, H.R.; Druffel, K.L.; Miglino, R.; van Schadewijk, A.R. Nucleotide sequence and genome organization of a member of a new and distinct Caulimovirus species from dahlia. Arch. Virol. 2008, 153, 2145–2148. [Google Scholar] [CrossRef]
- Schmidt, I.; Blanc, S.; Esperandieu, P.; Kuhl, G.; Devauchelle, G.; Louis, C.; Cerutti, M. Interaction between the aphid transmission factor and virus particles is a part of the molecular mechanism of cauliflower mosaic virus aphid transmission. Proc. Natl. Acad. Sci. USA 1994, 91, 8885–8889. [Google Scholar] [CrossRef]
- Leh, V.; Jacquot, E.; Geldreich, A.; Hermann, T.; Leclerc, D.; Cerutti, M.; Yot, P.; Keller, M.; Blanc, S. Aphid transmission of cauliflower mosaic virus requires the viral PIII protein. EMBO J. 1999, 18, 7077–7085. [Google Scholar] [CrossRef]
- Hohn, T. Plant pararetroviruses: Interactions of cauliflower mosaic virus with plants and insects. Curr. Opin. Virol. 2013, 3, 629–638. [Google Scholar] [CrossRef]
- Petrzik, K.; Beneš, V.; Mráz, I.; Honetšlegrová-Fránová, J.; Ansorge, W.; Špak, J. Strawberry Vein Banding Virus—Definitive Member of the Genus Caulimovirus. Virus Genes 1998, 16, 303–305. [Google Scholar] [CrossRef]
- Eid, S.; Almeyda, C.V.; Saar, D.E.; Druffel, K.L.; Pappu, H.R. Genomic characterization of pararetroviral sequences in wild Dahlia spp. in natural habitats. Arch. Virol. 2011, 156, 2079–2084. [Google Scholar] [CrossRef]
- Richins, R.D.; Scholthof, H.B.; Shepherd, R.J. Sequence of figwort mosaic virus DNA (caulimovirus group). Nucleic Acids Res. 1987, 15, 8451–8466. [Google Scholar] [CrossRef] [PubMed]
- Domier, L.L.; Steinlage, T.A.; Hobbs, H.A.; Wang, Y.; Herrera-Rodriguez, G.; Haudenshield, J.S.; McCoppin, N.K.; Hartman, G.L. Similarities in Seed and Aphid Transmission Among Soybean mosaic virus Isolates. Plant Dis. 2007, 91, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Brierley, P.; Smith, F.F. Some vectors, hosts, and properties of Dahlia mosaic virus. Plant Dis. Rep. 1950, 34, 363–367. [Google Scholar]
- Ferriol, I.; Rubio, L.; Perez-Panades, J.; Carbonell, E.A.; Davino, S.; Belliure, B. Transmissibility of Broad bean wilt virus 1 by aphids: Influence of virus accumulation in plants, virus genotype and aphid species. Ann. Appl. Biol. 2013, 162, 71–79. [Google Scholar] [CrossRef]
- Power, A.G. Insect transmission of plant viruses: A constraint on virus variability. Curr. Opin. Plant Biol. 2000, 3, 336–340. [Google Scholar] [CrossRef]
- Geijskes, R.J.; Braithwaite, K.S.; Dale, J.L.; Harding, R.M.; Smith, G.R. Sequence analysis of an Australian isolate of sugarcane bacilliform badnavirus. Arch. Virol. 2002, 147, 2393–2404. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Name | Amino Acid Identity (%) | ||||
|---|---|---|---|---|---|
| P1 | P2 | RdRp | LCP | SCP | |
| Broad bean wilt virus 1 | 44.83 | 44.99 1 | 51.15 | 46.51 1 | 41.06 |
| Broad bean wilt virus 2 | 43.77 | 43.01 | 49.22 | 45.90 | 40.00 |
| Cucurbit mild mosaic virus | 46.97 1 | 43.23 | 52.57 1 | 46.39 | 43.54 1 |
| Peach leaf pitting-associated virus | 43.87 | 25.33 | 48.79 | 26.96 | 19.77 |
| Gentian mosaic virus | 44.49 | 40.21 | 50.50 | 45.41 | 40.29 |
| Grapevine fabavirus | 36.64 | 24.36 | 45.43 | 20.52 | 17.49 |
| Lamium mild mosaic virus | 45.76 | 40.02 | 50.56 | 45.30 | 40.28 |
| Prunus virus F | 42.33 | 23.89 | 50.72 | 23.73 | 18.86 |
| Broad bean wilt virus 1 | 44.83 | 44.99 1 | 51.15 | 46.51 1 | 41.06 |
| Broad bean wilt virus 2 | 43.77 | 43.01 | 49.22 | 45.90 | 40.00 |
| Virus Name | Nucleotide Identities (%) | Amino Acid Identity (%) | |||||
|---|---|---|---|---|---|---|---|
| MP (P1) | ATF (P2) | DNAb (P3) | CP (P4) | PP (P5) | IB (P6) | ||
| LLDAV | 48.55 1 | 72.19 1 | 73.71 1 | 52.90 1 | 32.43 | 66.92 | 71.19 1 |
| CaMV | 30.44 | 56.44 | 58.03 | 30.84 | 34.58 | 68.28 1 | 32.72 |
| HLV | 24.85 | 55.04 | 60.13 | 37.41 | 36.13 | 65.05 | 32.58 |
| FMV | 23.32 | 44.30 | 34.61 | 30.43 | 35.07 | 65.43 | 18.16 |
| PVCV2 | 18.10 | 43.89 | 32.57 | 27.05 | 30.82 | 60.47 | 21.18 |
| AMMV | 24.67 | 46.70 | 38.16 | 38.79 | 30.17 | 63.29 | 28.23 |
| DaMV | 18.90 | 45.65 | 32.09 | 18.00 | 31.82 | 50.21 | 19.47 |
| MMV | 24.05 | 45.12 | 23.19 | 23.88 | 25.68 | 61.44 | 21.59 |
| CERV | 26.68 | 44.82 | 46.12 | 34.00 | 36.00 | 64.81 | 30.64 |
| SPuV | 24.54 | 45.38 | 38.16 | 33.65 | 36.38 1 | 64.09 | 25.94 |
| AnBSV | 25.19 | 42.23 | 41.54 | 30.43 | 31.27 | 66.30 | 27.91 |
| SVBV | 11.01 | 28.67 | 23.19 | 21.11 | 22.67 | 52.80 | 16.36 |
| Virus Name | Nucleotide Identities (%) | Amino Acid Identity (%) | |||||
|---|---|---|---|---|---|---|---|
| MP (P1) | ATF (P2) | DNAb (P3) | CP (P4) | PP (P5) | IB (P6) | ||
| FMV | 44.48 1 | 57.84 1 | 55.51 1 | 54.16 1 | 46.10 1 | 68.15 1 | 40.79 1 |
| CaMV | 13.00 | 51.50 | 31.41 | 30.85 | 29.80 | 56.03 | 19.71 |
| MMV | 16.27 | 48.83 | 41.87 | 38.02 | 36.90 | 59.86 | 27.19 |
| HLV | 14.52 | 45.71 | 29.27 | 33.21 | 31.08 | 55.26 | 20.49 |
| AMMV | 9.59 | 49.00 | 38.74 | 37.93 | 27.10 | 56.65 | 19.34 |
| DaMV | 18.20 | 45.03 | 38.15 | 44.17 | 35.97 | 53.53 | 27.65 |
| SPuV | 15.96 | 47.38 | 40.51 | 35.70 | 32.76 | 58.34 | 20.79 |
| LLDAV | 15.45 | 42.52 | 28.80 | 37.13 | 27.48 | 56.21 | 20.28 |
| PVCV1 | 18.10 | 43.89 | 32.57 | 27.05 | 30.82 | 60.47 | 21.18 |
| CERV | 5.74 | 45.47 | 29.77 | 32.09 | 28.84 | 55.92 | 21.03 |
| AnBSV | 15.55 | 37.80 | 33.92 | 29.23 | 24.83 | 55.51 | 16.96 |
| SVBV | 6.54 | 33.18 | 19.23 | None 2 | 24.38 | 46.51 | 14.50 |
| Virus Name | Amino Acid Identity (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | P4 | P5 | P7-1 | P7-2 | P8 | P9-1 | P9-2 | P10 | |
| MRDV | 45.06 | 35.17 1 | 32.27 | 21.63 | 25.18 1 | 25.04 | 4.74 | 18.92 | 26.42 1 | 19.84 1 | 20.14 |
| RBSDV | 43.97 | 35.00 | 32.24 | 21.70 | 13.68 | 25.39 | None 2 | 19.34 | 25.25 | 19.50 | 21.69 1 |
| SRBSDV | 45.90 1 | 34.97 | 32.53 | 19.38 | 21.99 | 25.23 | 5.37 1 | 18.08 | 24.75 | 18.20 | 20.72 |
| FDV | 44.74 | 34.96 | 32.56 1 | 19.71 | 19.87 | 24.17 | None 2 | 20.93 1 | 21.71 | 19.50 | 18.06 |
| MRCV | 45.41 | 35.08 | None 2 | 19.51 | 16.42 | 29.26 1 | None 2 | 18.18 | None 2 | 17.81 | 20.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, J.; Chen, Y.; Xie, Y.; Cao, M.; Fu, S.; Wu, J. The Identification of Viral Pathogens in a Physostegia virginiana Plant Using High-Throughput RNA Sequencing. Viruses 2023, 15, 1972. https://doi.org/10.3390/v15091972
Dong J, Chen Y, Xie Y, Cao M, Fu S, Wu J. The Identification of Viral Pathogens in a Physostegia virginiana Plant Using High-Throughput RNA Sequencing. Viruses. 2023; 15(9):1972. https://doi.org/10.3390/v15091972
Chicago/Turabian StyleDong, Jinxi, Yuanling Chen, Yi Xie, Mengji Cao, Shuai Fu, and Jianxiang Wu. 2023. "The Identification of Viral Pathogens in a Physostegia virginiana Plant Using High-Throughput RNA Sequencing" Viruses 15, no. 9: 1972. https://doi.org/10.3390/v15091972
APA StyleDong, J., Chen, Y., Xie, Y., Cao, M., Fu, S., & Wu, J. (2023). The Identification of Viral Pathogens in a Physostegia virginiana Plant Using High-Throughput RNA Sequencing. Viruses, 15(9), 1972. https://doi.org/10.3390/v15091972