

Spreading of the High-Pathogenicity Avian Influenza (H5N1) Virus of Clade 2.3.4.4b into Uruguay

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statements
2.2. Sample Collections
2.3. AIV Detection
2.4. HA Subtyping
2.5. RNA Extraction and Illumina Sequencing
2.6. Genome Assembly and Annotation
2.7. Phylogenetic Analysis
3. Results
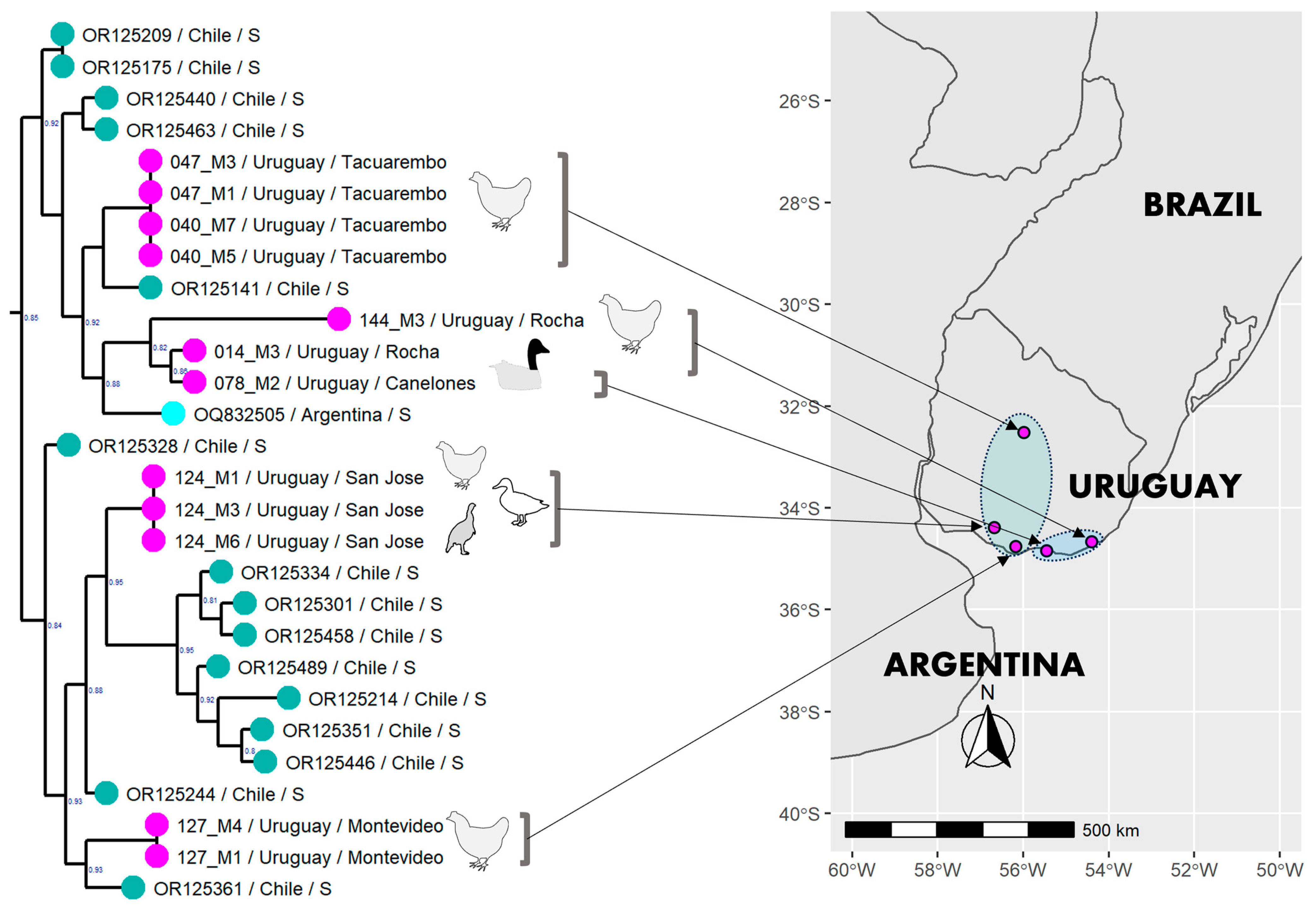
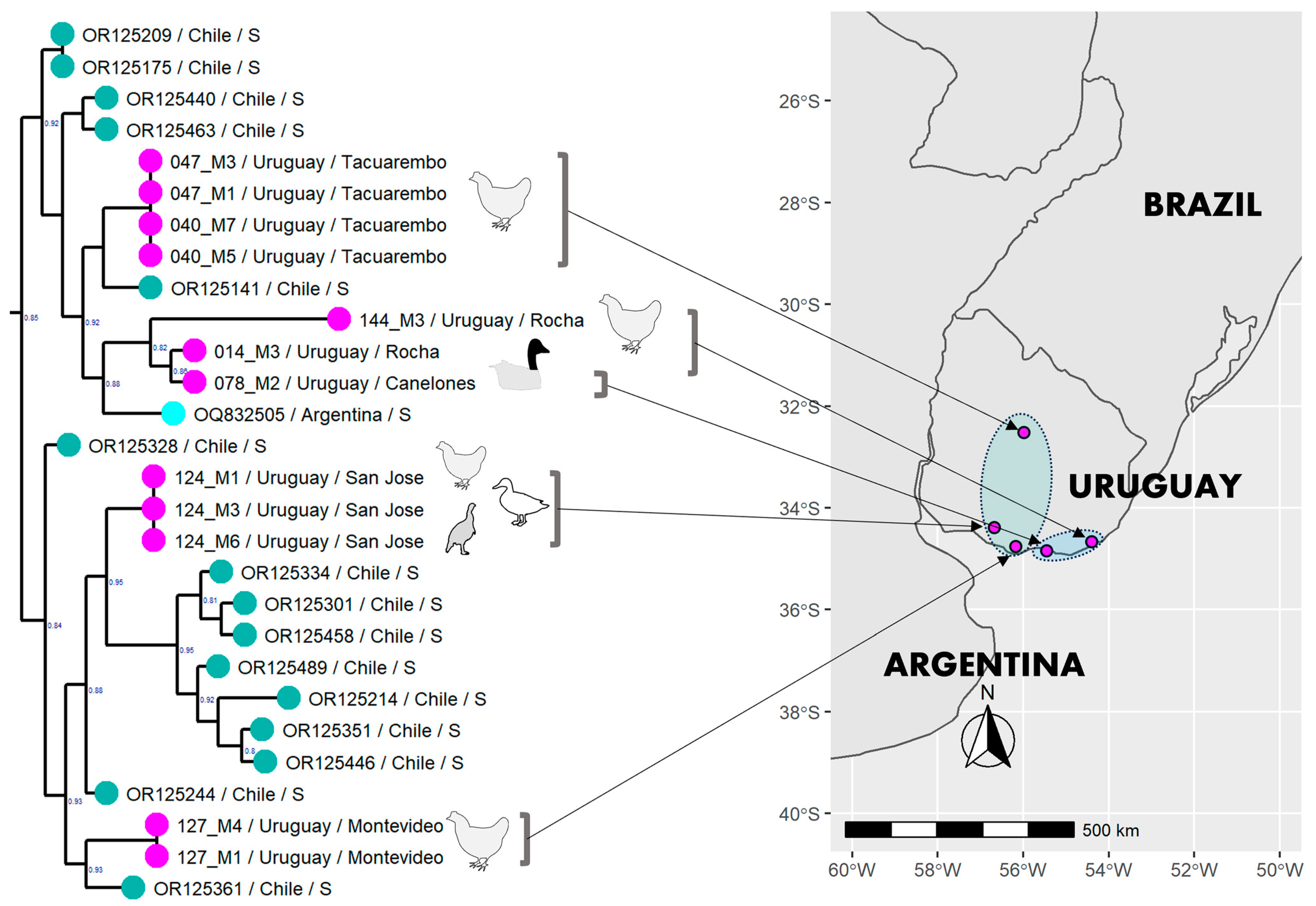
3.1. Description of the Outbreaks
3.2. Genome Sequence Comparison
3.3. HA Sequence Comparison
3.4. HPAIV Cleavage Site
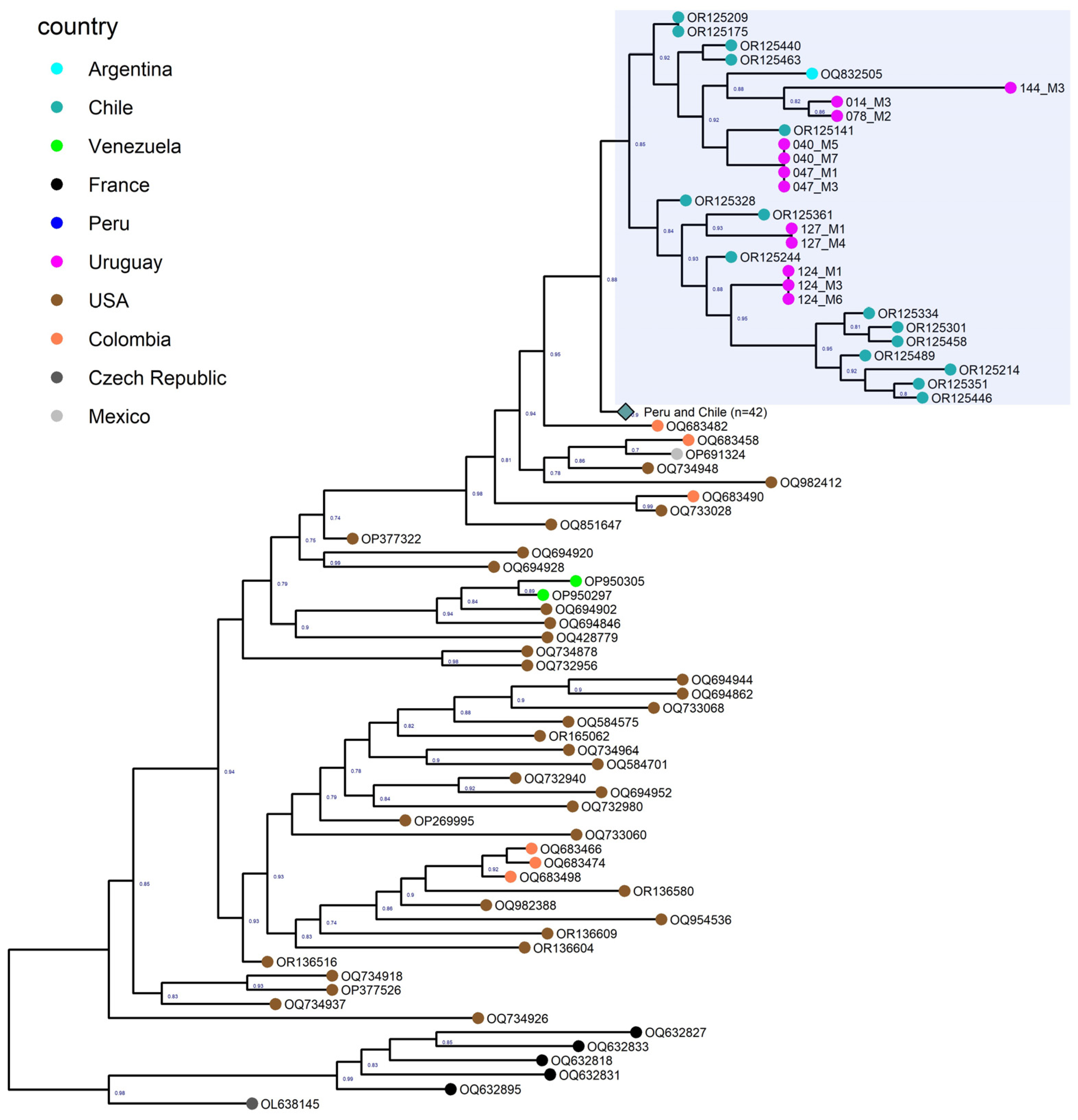
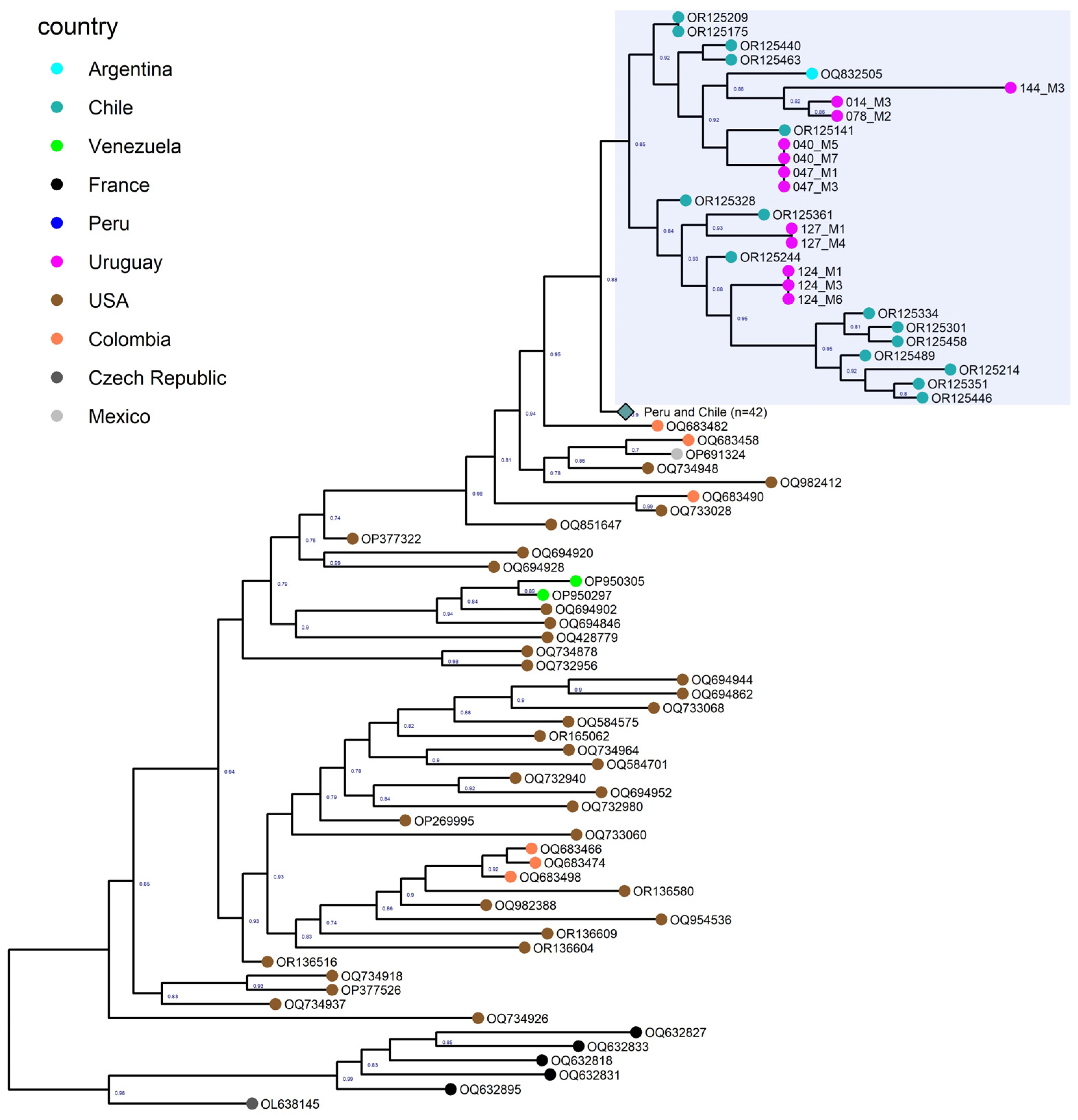
3.5. Phylogenetic Analysis of North and South American Strains
3.6. Phylogenetic Analysis of the Uruguayan Strains-Containing Clade
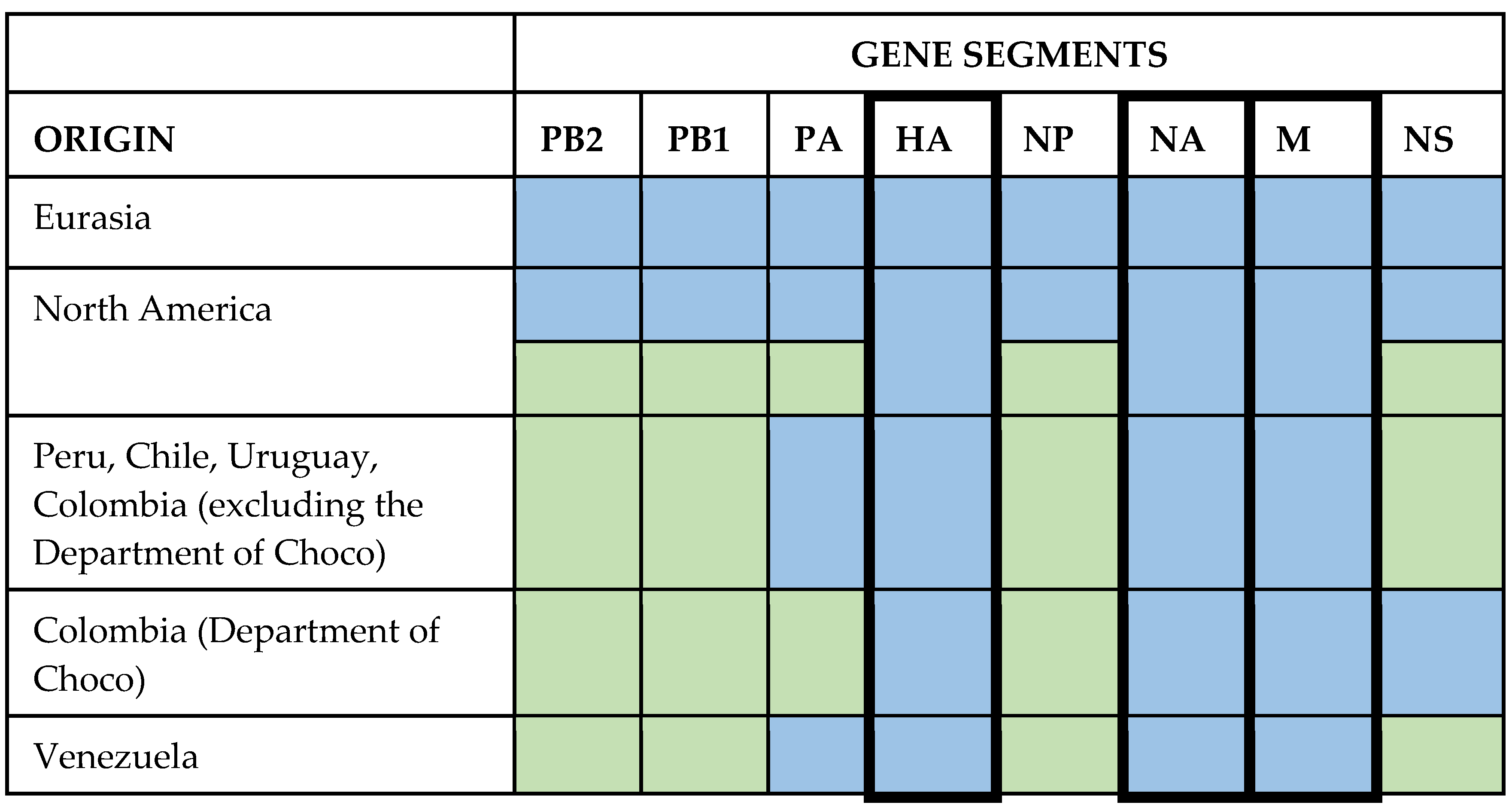
3.7. Other Segments Comparison
4. Discussion
4.1. Detection and Genomic Characterization
4.2. Local Divergence
4.3. Pathogenicity
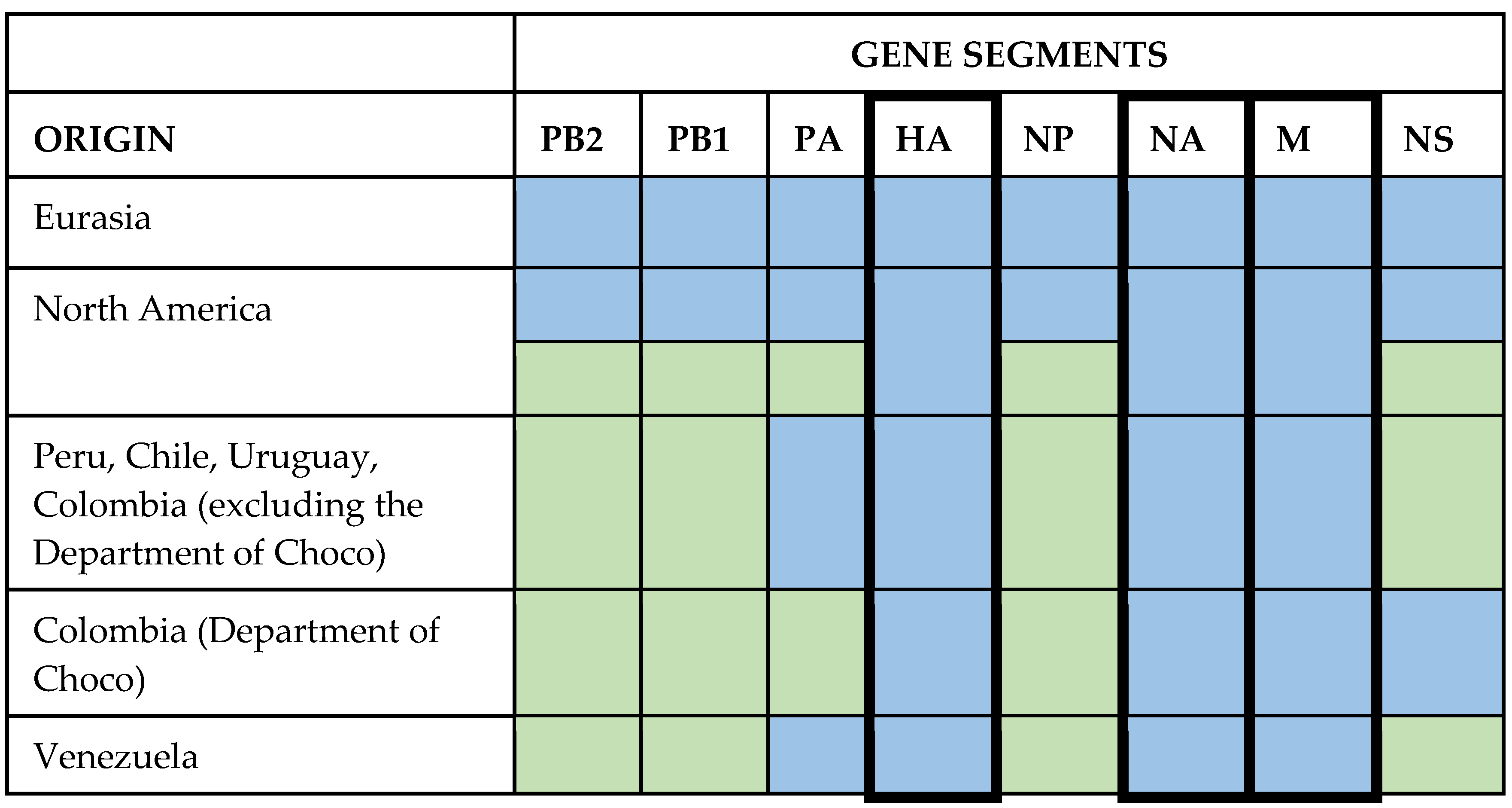
4.4. Reassortments in American Strains
4.5. Spreading in South America through the Pacific Flyway
4.6. Avian Influenza Poses a Significant Risk to the Poultry Sector in South America
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Spackman, E. A Brief Introduction to the Avian Influenza Virus. In Avian Influenza Virus; Humana Press: Totowa, NJ, USA, 2008; pp. 1–6. [Google Scholar]
- Mostafa, A.; Abdelwhab, E.M.; Mettenleiter, T.C.; Pleschka, S. Zoonotic Potential of Influenza A Viruses: A Comprehensive Overview. Viruses 2018, 10, 497. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2008, 16, e3000003. [Google Scholar] [CrossRef]
- Kuiken, T. Is low pathogenic avian influenza virus virulent for wild waterbirds? Proc. R. Soc. B Biol. Sci. 2013, 280, 20130990. [Google Scholar] [CrossRef]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenström, J.; Osterhaus, A.D.; Fouchier, R.A. Global Patterns of Influenza A Virus in Wild Birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef]
- Webster, R.G.; Yakhno, M.; Hinshaw, V.S.; Bean, W.J.; Murti, K.C. Intestinal influenza: Replication and characterization of influenza viruses in ducks. Virology 1978, 84, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.F. Lessons from Emergence of A/Goose/Guangdong/1996-Like H5N1 Highly Pathogenic Avian Influenza Viruses and Recent Influenza Surveillance Efforts in Southern China. Zoonoses Public Health 2012, 59, 32–42. [Google Scholar] [CrossRef]
- Xu, X.; Subbarao, K.; Cox, N.J.; Guo, Y. Genetic Characterization of the Pathogenic Influenza A/Goose/Guangdong/1/96 (H5N1) Virus: Similarity of Its Hemagglutinin Gene to Those of H5N1 Viruses from the 1997 Outbreaks in Hong Kong. Virology 1999, 261, 15–19. [Google Scholar] [CrossRef]
- Hill, N.J.; Hussein, I.T.; Davis, K.R.; Ma, E.J.; Spivey, T.J.; Ramey, A.M.; Puryear, W.B.; Das, S.R.; Halpin, R.A.; Lin, X.; et al. Reassortment of influenza a viruses in wild birds in alaska before H5 clade 2.3.4.4 outbreaks. Emerg. Infect. Dis. 2017, 23, 654–657. [Google Scholar] [CrossRef]
- Shao, W.; Li, X.; Goraya, M.U.; Wang, S.; Chen, J.-L. Evolution of Influenza A Virus by Mutation and Re-Assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef]
- Dhingra, M.S.; Artois, J.; Robinson, T.P.; Linard, C.; Chaiban, C.; Xenarios, I.; Engler, R.; Liechti, R.; Kuznetsov, D.; Xiao, X.; et al. Global mapping of highly pathogenic avian influenza H5N1 and H5Nx clade 2.3.4.4 viruses with spatial cross-validation. eLife 2016, 5, e19571. [Google Scholar] [CrossRef] [PubMed]
- Antigua, K.J.C.; Choi, W.-S.; Baek, Y.H.; Song, M.-S. The Emergence and Decennary Distribution of Clade 2.3.4.4 HPAI H5Nx. Microorganisms 2019, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.S.; Banyard, A.C.; Whittard, E.; Karibayev, T.; Al Kafagi, T.; Chvala, I.; Byrne, A.; Meruyert, S.; King, J.; Harder, T.; et al. Emergence and spread of novel H5N8, H5N5 and H5N1 clade 2.3.4.4 highly pathogenic avian influenza in 2020. Emerg. Microbes Infect. 2021, 10, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Shi, J.; Cui, P.; Yan, C.; Zhang, Y.; Wang, C.; Zhang, Y.; Xing, X.; Zeng, X.; Liu, L.; et al. Novel H5N6 reassortants bearing the clade 2.3.4.4b HA gene of H5N8 virus have been detected in poultry and caused multiple human infections in China. Emerg. Microbes Infect. 2022, 11, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Bevins, S.N.; Shriner, S.A.; Cumbee, J.C.; Dilione, K.E.; Douglass, K.E.; Ellis, J.W.; Killian, M.L.; Torchetti, M.K.; Lenoch, J.B. Intercontinental Movement of Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4 Virus to the United States, 2021. Emerg. Infect. Dis. 2022, 28, 1006–1011. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; Terregino, C.; Aznar, I.; Guajardo, I.M.; et al. Avian influenza overview December 2021–March 2022. EFSA J. 2022, 20, e07289. [Google Scholar] [CrossRef]
- Caliendo, V.; Lewis, N.S.; Pohlmann, A.; Baillie, S.R.; Banyard, A.C.; Beer, M.; Brown, I.H.; Fouchier, R.A.M.; Hansen, R.D.E.; Lameris, T.K.; et al. Transatlantic spread of highly pathogenic avian influenza H5N1 by wild birds from Europe to North America in 2021. Sci. Rep. 2022, 12, 11729. [Google Scholar] [CrossRef]
- Bruno, A.; Alfaro-Núñez, A.; de Mora, D.; Armas, R.; Olmedo, M.; Garcés, J.; Muñoz-López, G.; Garcia-Bereguiain, M.A. First case of human infection with highly pathogenic H5 avian influenza a virus in South America: A new zoonotic pandemic threat for 2023? J. Travel Med. 2023, taad032. [Google Scholar] [CrossRef] [PubMed]
- Puryear, W.; Sawatzki, K.; Hill, N.; Foss, A.; Stone, J.J.; Doughty, L.; Walk, D.; Gilbert, K.; Murray, M.; Cox, E.; et al. Highly Pathogenic Avian Influenza A(H5N1) Virus Outbreak in New England Seals, United States. Emerg. Infect. Dis. 2023, 29, 786–791. [Google Scholar] [CrossRef]
- Günther, A.; Krone, O.; Svansson, V.; Pohlmann, A.; King, J.; Hallgrimsson, G.T.; Skarphéðinsson, K.H.; Sigurðardóttir, H.; Jónsson, S.R.; Beer, M.; et al. Iceland as Stepping Stone for Spread of Highly Pathogenic Avian Influenza Virus between Europe and North America. Emerg. Infect. Dis. 2022, 28, 2383–2388. [Google Scholar] [CrossRef]
- Ariyama, N.; Pardo-Roa, C.; Munoz, G.; Aguayo, C.; Avila, C.; Mathieu, C.; Brito, B.; Medina, R.; Johow, M.; Neira-Ramirez, V. Emergence and rapid dissemination of highly pathogenic avian influenza virus H5N1 clade 2.3.4.4b in wild birds, Chile. bioRxiv 2023. preprint. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Mirinaviciute, G.; Niqueux, É.; Stahl, K.; Staubach, C.; Terregino, C.; et al. Avian influenza overview December 2022–March 2023. EFSA J. 2023, 21, e07917. [Google Scholar] [CrossRef] [PubMed]
- Gamarra-Toledo, V.; Plaza, P.I.; Gutiérrez, R.; Luyo, P.; Hernani, L.; Angulo, F.; Lambertucci, S.A. Avian flu threatens Neotropical birds. Science 2023, 379, 246. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; George, K.S.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-Reaction Genomic Amplification Accelerates Sequencing and Vaccine Production for Classical and Swine Origin Human Influenza A Viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Ruiz-Saenz, J.; Martinez-Gutierrez, M.; Pujol, F.H. Multiple introductions of highly pathogenic avian influenza H5N1 clade 2.3.4.4b into South America. Travel Med. Infect. Dis. 2023, 53, 102591. [Google Scholar] [CrossRef]
- Ellis, T.M.; Bousfield, R.B.; Bissett, L.A.; Dyrting, K.C.; Luk, G.S.M.; Tsim, S.T.; Sturm-Ramirez, K.; Webster, R.G.; Guan, Y.; Peiris, J.S.M. Investigation of outbreaks of highly pathogenic H5N1 avian influenza in waterfowl and wild birds in Hong Kong in late 2002. Avian Pathol. 2004, 33, 492–505. [Google Scholar] [CrossRef]
- Karawita, A.C.; Cheng, Y.; Chew, K.Y.; Challagulla, A.; Kraus, R.; Mueller, R.C.; Tong, M.Z.W.; Hulme, K.D.; Bielefeldt-Ohmann, H.; Steele, L.E.; et al. The swan genome and transcriptome, it is not all black and white. Genome Biol. 2023, 24, 13. [Google Scholar] [CrossRef] [PubMed]
- Castro-Sanguinetti, G.; Gonzalez-Veliz, R.; Callupe-Leyva, A.; Apaza-Chiara, A.; Jara, J.; Silva, W.; Icochea, E.; More-Bayona, J. Circulation of highly pathogenic avian influenza virus H5N1 clade 2.3.4.4b in highly diverse wild bird species from Peru. Res. Sq. 2023. preprint. [Google Scholar]
- Leguia, M.; Garcia-Glaessner, A.; Muñoz-Saavedra, B.; Juarez, D.; Barrera, P.; Calvo-Mac, C.; Jara, J.; Silva, W.; Ploog, K.; Amaro, L.; et al. Highly pathogenic avian influenza A (H5N1) in marine mammals and seabirds in Peru. bioRxiv 2023. preprint. [Google Scholar] [CrossRef]
- Jimenez-Bluhm, P.; Siegers, J.Y.; Tan, S.; Sharp, B.; Freiden, P.; Johow, M.; Orozco, K.; Ruiz, S.; Baumberger, C.; Galdames, P.; et al. Detection and Phylogenetic Analysis of Highly Pathogenic A/H5N1 Avian Influenza Clade 2.3.4.4b Virus in Chile, 2022. Emerg. Microbes Infect. 2023, 12, 2220569. [Google Scholar] [CrossRef]
- Scholtissek, C.; Quack, G.; Klenk, H.D.; Webster, R.G. How to overcome resistance of influenza A viruses against adamantane derivatives. Antivir. Res. 1998, 37, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Treanor, J.J.; Hayden, F.G.; Vrooman, P.S.; Barbarash, R.; Bettis, R.; Riff, D.; Singh, S.; Kinnersley, N.; Ward, P.; Mills, R.G.; et al. Efficacy and Safety of the Oral Neuraminidase Inhibitor Oseltamivir in Treating Acute Influenza. JAMA 2000, 283, 1016. [Google Scholar] [CrossRef]
- Kandeil, A.; Patton, C.; Jones, J.C.; Jeevan, T.; Harrington, W.N.; Trifkovic, S.; Seiler, J.P.; Fabrizio, T.; Woodard, K.; Turner, J.C.; et al. Rapid evolution of A(H5N1) influenza viruses after intercontinental spread to North America. Nat. Commun. 2023, 14, 3082. [Google Scholar] [CrossRef]
- Xie, R.; Edwards, K.M.; Wille, M.; Wei, X.; Wong, S.S.; Zanin, M.; El-Shesheny, R.; Ducatez, M.; Poon, L.L.; Kayali, G.; et al. The episodic resurgence of highly pathogenic avian influenza H5 virus. bioRxiv 2022. preprint. [Google Scholar] [CrossRef]
- PAHO/WHO. Epidemiological Update Outbreaks of Avian Influenza Caused by Influenza A(H5N1) in the Region of the Americas; PAHO/WHO: Washington, DC, USA, 2023. [Google Scholar]
- Capllonch, P. Un panorama de las migraciones de aves en Argentina. El Hornero 2018, 33, 1–17. [Google Scholar] [CrossRef]
- Schlatter, R.P.; Navarro, R.A.; Corti, P. Effects of El Nino Southern Oscillation on Numbers of Black-Necked Swans at Rio Cruces Sanctuary, Chile. Waterbirds 2002, 25, 114–122. [Google Scholar]
- Rees, E.C.; Clausen, P.; Coleman, J.T. Conservation status of the world’s swan populations, Cygnus sp. and Coscoroba sp.: A review of current trends and gaps in knowledge. Wildfowl 2019, 5, 37–72. [Google Scholar]
- Teitelbaum, C.S.; Casazza, M.L.; McDuie, F.; De La Cruz, S.E.W.; Overton, C.T.; Hall, L.A.; Matchett, E.L.; Ackerman, J.T.; Sullivan, J.D.; Ramey, A.M.; et al. Waterfowl recently infected with low pathogenic avian influenza exhibit reduced local movement and delayed migration. Ecosphere 2023, 14, e4432. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Date | Sample | Origin | Species | RT-qPCR AIV Matrix (Cq) | Accession |
|---|---|---|---|---|---|
| 18 February 2023 | 014_M3 | Rocha (Laguna de Garzón) | Wild, black-necked swan | 26.5–27.8 | OR381584-OR381591 |
| 3 March 2023 | 040_M5 | Tacuarembó | Backyard chicken | 25.2 | OR381592-OR381599 |
| 3 March 2023 | 040_M7 | Tacuarembó | Backyard chicken | 22.6 | OR381600-OR381607 |
| 6 March 2023 | 047_M1 | Tacuarembó | Backyard chicken | 18.2 | OR381608-OR381615 |
| 6 March 2023 | 047_M3 | Tacuarembó | Backyard chicken | 18.9 | OR381616-OR381623 |
| 15 March 2023 | 078_M2 | Canelones (Solymar) | Wild, black-necked swan | 25 | OR381672-OR381679 |
| 16 April 2023 | 124_M1 | San José | Backyard chicken | 20.6 | OR381624-OR381631 |
| 16 April 2023 | 124_M3 | San José | Backyard duck | 19.5 | OR381656-OR381663 |
| 16 April 2023 | 124_M6 | San José | Backyard turkey | 29.1 | OR381664-OR381671 |
| 17 April 2023 | 127_M1 | Montevideo | Backyard chicken | 22.1–27.5 | OR381632-OR381639 |
| 17 April 2023 | 127_M4 | Montevideo | Backyard chicken | 22.1–27.5 | OR381640-OR381647 |
| 3 May 2023 | 144_M3 | Rocha | Backyard chicken | 15.8 | OR381648-OR381655 |
| PB2 | PB1 | PA | HA | NP | NA | M | NS1/NEP | Total | |
|---|---|---|---|---|---|---|---|---|---|
| Nt (CDS) | 2213 | 2274 | 2151 | 1704 (18 aa signal peptide) | 1497 | 1410 | 982 | 838 (NS) 693/366 | 13.069 |
| SNP | 26 | 27 | 21 | 20 | 14 | 14 | 7 | 13 | 142 |
| Syn | 17 | 21 | 11 | 13 | 10 | 9 | 2 | 6 | 89 |
| Non-syn | 9 | 6 | 10 | 7 | 4 | 5 | 5 | 7 | 53 |
| Sequences with nnns (length) | 014_M3 (294) | 014_M3 (427 nt) | 014_M3 (22 nt) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marandino, A.; Tomás, G.; Panzera, Y.; Leizagoyen, C.; Pérez, R.; Bassetti, L.; Negro, R.; Rodríguez, S.; Pérez, R. Spreading of the High-Pathogenicity Avian Influenza (H5N1) Virus of Clade 2.3.4.4b into Uruguay. Viruses 2023, 15, 1906. https://doi.org/10.3390/v15091906
Marandino A, Tomás G, Panzera Y, Leizagoyen C, Pérez R, Bassetti L, Negro R, Rodríguez S, Pérez R. Spreading of the High-Pathogenicity Avian Influenza (H5N1) Virus of Clade 2.3.4.4b into Uruguay. Viruses. 2023; 15(9):1906. https://doi.org/10.3390/v15091906
Chicago/Turabian StyleMarandino, Ana, Gonzalo Tomás, Yanina Panzera, Carmen Leizagoyen, Ramiro Pérez, Lucía Bassetti, Raúl Negro, Sirley Rodríguez, and Ruben Pérez. 2023. "Spreading of the High-Pathogenicity Avian Influenza (H5N1) Virus of Clade 2.3.4.4b into Uruguay" Viruses 15, no. 9: 1906. https://doi.org/10.3390/v15091906
APA StyleMarandino, A., Tomás, G., Panzera, Y., Leizagoyen, C., Pérez, R., Bassetti, L., Negro, R., Rodríguez, S., & Pérez, R. (2023). Spreading of the High-Pathogenicity Avian Influenza (H5N1) Virus of Clade 2.3.4.4b into Uruguay. Viruses, 15(9), 1906. https://doi.org/10.3390/v15091906