
Immune Response to Chikungunya Virus: Sex as a Biological Variable and Implications for Natural Delivery via the Mosquito


Abstract
1. Introduction
2. Innate Immune Response to CHIKV
2.1. Role Type I Interferons during CHIKV Infection
2.2. Innate Immune Cells during CHIKV Infection
2.3. Role of Pattern Recognition Receptors in Innate Immunity to CHIKV
3. Adaptive Immune Response to CHIKV
4. Sex and Age as Biological Variables in CHIKV Immune Response
4.1. Sex as a Biological Variable
4.2. Age as a Biological Variable
5. Contribution of Mosquito Saliva to CHIKV Pathogenesis

{kind=link}
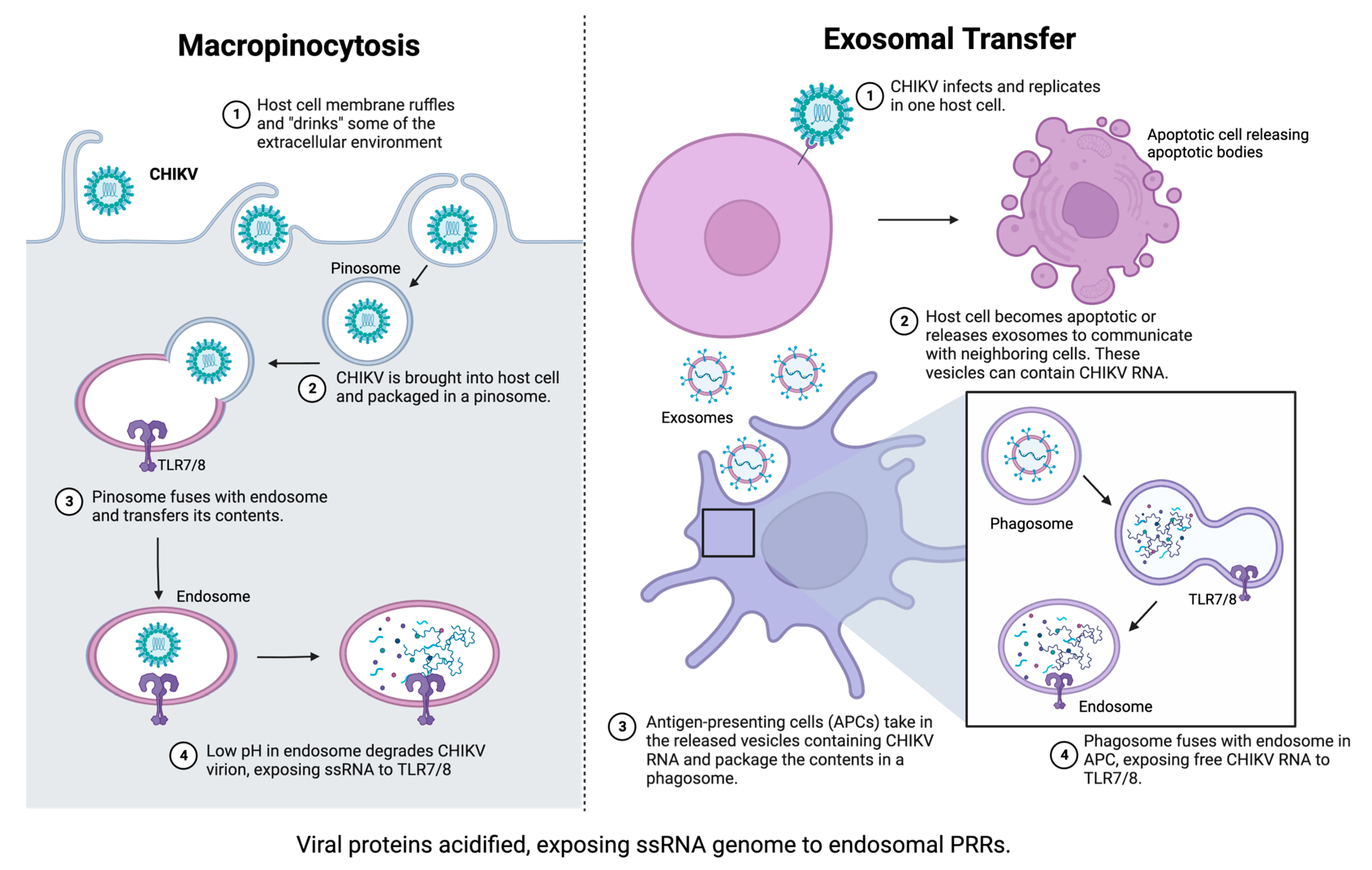{kind=link}
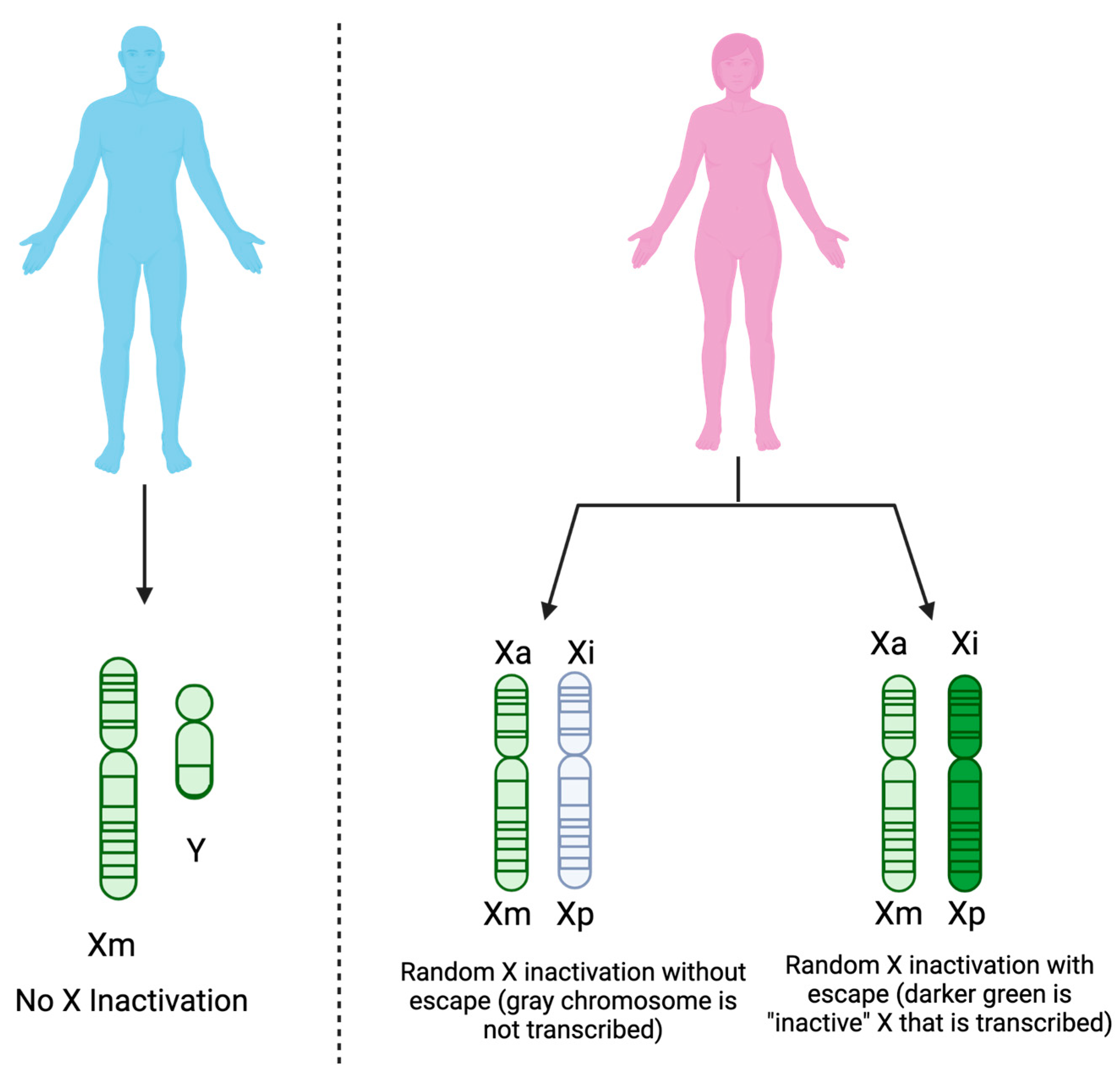{kind=link}
| Host Species | Virus (Strain If Provided) | Vector Species + Method | Impact on Host Response | References |
|---|---|---|---|---|
| Swiss albino pups | CHIKV (DRDE-06) | Ae. aegypti bite + SubQ needle inoc. | increased morbidity and mortality | [71] |
| higher skin viral RNA load and viremia | ||||
| more extensive viral dissemination | ||||
| dysregulated cytokine release | ||||
| enhanced cellular infiltration | ||||
| BALB/c mice | SFV4 (pCMV-SFV4) | Ae. aegypti bite + SubQ needle inoc. | extensive edema (viral inoculum retained at bite site) | [73] |
| transient increase in skin neutrophils | ||||
| CD-1 mice (2 weeks) | CHIKV (LR 5’GFP) | Ae aegypti bite inoculation | suppression of Th1 cytokines, enhancement of Th2 cytokines | [74] |
| downregulation of TLR3 expression | ||||
| downregulation of IFN-gamma expression | ||||
| Human dermal fibroblasts | CHIKV (LR2006_OPY1) | Ae. aegypti saliva | decreased ISG expression | [75] |
| downregulation of STAT2 and pSTAT2 | ||||
| increased CHIKV titer over time | ||||
| Hu-NSG Mice | CHIKV (37997) | Ae. aegypti bite inoculation | exhibit more human-like CHIKV symptomology | [76] |
| increased circulating CHIKV RNA | ||||
| increased dissemination over needle-inoculation | ||||
| differential cell recruitment compared to needle inoculation | ||||
| Human Keratinocytes | DENV | recombinant Aedes salivary proteins (putative 34 kDa) | (1) decreased IFN-I mRNA expression | [71,77] |
| (2) decreased IRF3 and IRF7 mRNA expression | ||||
| (3) suppressed antimicrobial peptide expression | ||||
| (4) increased viral RNA titers |
6. Gaps in Knowledge
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clarence Peters, J.J.; Dalrymple, J.M. Chapter 26: Alphaviruses, 2nd ed.; Fields, B.N., Ed.; Raven Press Ltd: New York, NY, USA, 1990. [Google Scholar]
- Schwartz, O.; Albert, M.L. Biology and Pathogenesis of Chikungunya Virus. Nat. Rev. Microbiol. 2010, 8, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Galán-Huerta, K.A.; Rivas-Estilla, A.M.; Fernández-Salas, I.; Farfan-Ale, J.A.; Ramos-Jiménez, J. Chikungunya Virus: A General Overview. Med. Univ. 2015, 17, 175–183. [Google Scholar] [CrossRef]
- Ryan, S.J.; Carlson, C.J.; Mordecai, E.A.; Johnson, L.R. Global Expansion and Redistribution of Aedes-Borne Virus Transmission Risk with Climate Change. PLoS Negl. Trop. Dis. 2019, 13, e0007213. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, L.; Scott, T.W.; Gubler, D.J. Consequences of the Expanding Global Distribution of Aedes Albopictus for Dengue Virus Transmission. PLoS Negl. Trop. Dis. 2010, 4, e646. [Google Scholar] [CrossRef]
- CDC Yellow Book 2024. Available online: https://wwwnc.cdc.gov/travel/yellowbook/2024/infections-diseases/chikungunya (accessed on 9 February 2023).
- Gérardin, P.; Barau, G.; Michault, A.; Bintner, M.; Randrianaivo, H.; Choker, G.; Lenglet, Y.; Touret, Y.; Bouveret, A.; Grivard, P.; et al. Multidisciplinary Prospective Study of Mother-to-Child Chikungunya Virus Infections on the Island of La Réunion. PLoS Med. 2008, 5, e60. [Google Scholar] [CrossRef] [PubMed]
- Economopoulou, A.; Dominguez, M.; Helynck, B.; Sissoko, D.; Wichmann, O.; Quenel, P.; Germonneau, P.; Quatresous, I. Atypical Chikungunya Virus Infections: Clinical Manifestations, Mortality and Risk Factors for Severe Disease during the 2005–2006 Outbreak on Réunion. Epidemiol. Infect. 2009, 137, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, M.F.S.d.; Silva, A.P.d.S.C.; Lacerda, H.R. Factors Associated with Death from Dengue and Chikungunya Virus Infection during an Epidemic Period in Northeast Brazil: A Retrospective Cohort Study. Rev. Soc. Bras. Med. Trop. 2023, 56, e0030. [Google Scholar] [CrossRef]
- Ramachandran, V.; Malaisamy, M.; Ponnaiah, M.; Kaliaperuaml, K.; Vadivoo, S.; Gupte, M.D. Impact of Chikungunya on Health Related Quality of Life Chennai, South India. PLoS ONE 2012, 7, e51519. [Google Scholar] [CrossRef]
- Vidal, O.M.; Acosta-Reyes, J.; Padilla, J.; Navarro-Lechuga, E.; Bravo, E.; Viasus, D.; Arcos-Burgos, M.; Vélez, J.I. Chikungunya Outbreak (2015) in the Colombian Caribbean: Latent Classes and Gender Differences in Virus Infection. PLoS Negl. Trop. Dis. 2020, 14, e0008281. [Google Scholar] [CrossRef]
- Zingman, M.A.; Paulino, A.T.; Payano, M.P. Clinical Manifestations of Chikungunya among University Professors and Staff in Santo Domingo, the Dominican Republic. Rev. Panam. De Salud Pública 2017, 41, e64. [Google Scholar] [CrossRef]
- Sourisseau, M.; Schilte, C.; Casartelli, N.; Trouillet, C.; Guivel-Benhassine, F.; Rudnicka, D.; Sol-Foulon, N.; Roux, K.L.; Prevost, M.-C.; Fsihi, H.; et al. Characterization of Reemerging Chikungunya Virus. PLoS Pathog. 2007, 3, e89. [Google Scholar] [CrossRef]
- Schilte, C.; Couderc, T.; Chretien, F.; Sourisseau, M.; Gangneux, N.; Guivel-Benhassine, F.; Kraxner, A.; Tschopp, J.; Higgs, S.; Michault, A.; et al. Type I IFN Controls Chikungunya Virus via Its Action on Nonhematopoietic Cells. J. Exp. Med. 2010, 207, 429–442. [Google Scholar] [CrossRef]
- Lester, S.N.; Li, K. Toll-like Receptors in Antiviral Innate Immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar]
- Izumida, M.; Hayashi, H.; Tanaka, A.; Kubo, Y. Cathepsin B Protease Facilitates Chikungunya Virus Envelope Protein-Mediated Infection via Endocytosis or Macropinocytosis. Viruses 2020, 12, 722. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I Interferons in Infectious Disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar]
- WAGNER, R.R. Inhibition of Interferon Biosynthesis by Actinomycin D. Nature 1964, 204, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.M. Role of Interferon in Viral Interference. Nature 1964, 201, 848–849. [Google Scholar] [CrossRef] [PubMed]
- Her, Z.; Malleret, B.; Chan, M.; Ong, E.K.S.; Wong, S.-C.; Kwek, D.J.C.; Tolou, H.; Lin, R.T.P.; Tambyah, P.A.; Rénia, L.; et al. Active Infection of Human Blood Monocytes by Chikungunya Virus Triggers an Innate Immune Response. J. Immunol. 2010, 184, 5903–5913. [Google Scholar] [CrossRef] [PubMed]
- Rudd, P.A.; Wilson, J.; Gardner, J.; Larcher, T.; Babarit, C.; Le, T.T.; Anraku, I.; Kumagai, Y.; Loo, Y.-M.; Gale, M.; et al. Interferon Response Factors 3 and 7 Protect against Chikungunya Virus Hemorrhagic Fever and Shock. J. Virol. 2012, 86, 9888–9898. [Google Scholar] [CrossRef]
- Gardner, J.; Anraku, I.; Le, T.T.; Larcher, T.; Major, L.; Roques, P.; Schroder, W.A.; Higgs, S.; Suhrbier, A. Chikungunya Virus Arthritis in Adult Wild-Type Mice. J. Virol. 2010, 84, 8021–8032. [Google Scholar] [CrossRef]
- Phuklia, W.; Kasisith, J.; Modhiran, N.; Rodpai, E.; Thannagith, M.; Thongsakulprasert, T.; Smith, D.R.; Ubol, S. Osteoclastogenesis Induced by CHIKV-Infected Fibroblast-like Synoviocytes: A Possible Interplay between Synoviocytes and Monocytes/Macrophages in CHIKV-Induced Arthralgia/Arthritis. Virus Res. 2013, 177, 179–188. [Google Scholar] [CrossRef]
- Tanabe, I.S.B.; Tanabe, E.L.L.; Santos, E.C.; Martins, W.V.; Araújo, I.M.T.C.; Cavalcante, M.C.A.; Lima, A.R.V.; Câmara, N.O.S.; Anderson, L.; Yunusov, D.; et al. Cellular and Molecular Immune Response to Chikungunya Virus Infection. Front. Cell. Infect. Microbiol. 2018, 8, 345. [Google Scholar]
- Hoarau, J.-J.; Jaffar Bandjee, M.-C.; Krejbich Trotot, P.; Das, T.; Li-Pat-Yuen, G.; Dassa, B.; Denizot, M.; Guichard, E.; Ribera, A.; Henni, T.; et al. Persistent Chronic Inflammation and Infection by Chikungunya Arthritogenic Alphavirus in Spite of a Robust Host Immune Response. J. Immunol. 2010, 184, 5914–5927. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.A.; Lu, L.; da Rosa, A.P.; Xiao, S.Y.; Tesh, R.B. An animal model for studying the pathogenesis of chikungunya virus infection. Am. J. Trop. Med. Hyg. 2008, 79, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Dupuis-Maguiraga, L.; Noret, M.; Brun, S.; Grand, R.L.; Gras, G.; Roques, P. Chikungunya Disease: Infection-Associated Markers from the Acute to the Chronic Phase of Arbovirus-Induced Arthralgia. PLoS Negl. Trop. Dis. 2012, 6, e1446. [Google Scholar] [CrossRef] [PubMed]
- Labadie, K.; Larcher, T.; Joubert, C.; Mannioui, A.; Delache, B.; Brochard, P.; Guigand, L.; Dubreil, L.; Lebon, P.; Verrier, B.; et al. Chikungunya Disease in Nonhuman Primates Involves Long-Term Viral Persistence in Macrophages. J. Clin. Investig. 2010, 120, 894–906. [Google Scholar] [CrossRef]
- Webster, B.; Werneke, S.W.; Zafirova, B.; Bastien This, S.; Verin Colé On, S.; Dé Cembre, E.; Paidassi, H.; Bouvier, I.; Joubert, P.-E.; Duffy, D.; et al. Plasmacytoid Dendritic Cells Control Dengue and Chikungunya Virus Infections via IRF7-Regulated Interferon Responses. Elife 2018, 7, e34273. [Google Scholar] [CrossRef]
- Hong, J.P.; McCarthy, M.K.; Davenport, B.J.; Morrison, T.E.; Diamond, M.S. Clearance of Chikungunya Virus Infection in Lymphoid Tissues Is Promoted by Treatment with an Agonistic Anti-CD137 Antibody. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal. Transduct. Target. Ther. 2021, 6, 291. [Google Scholar]
- Her, Z.; Teng, T.; Tan, J.J.; Teo, T.; Kam, Y.; Lum, F.; Lee, W.W.; Gabriel, C.; Melchiotti, R.; Andiappan, A.K.; et al. Loss of TLR3 Aggravates CHIKV Replication and Pathology Due to an Altered Virus-specific Neutralizing Antibody Response. EMBO Mol. Med. 2015, 7, 24–41. [Google Scholar] [CrossRef]
- Sanchez David, R.Y.; Combredet, C.; Sismeiro, O.; Dillies, M.-A.; Jagla, B.; Coppé, J.-Y.; Mura, M.; Guerbois Galla, M.; Despres, P.; dé ric Tangy, F.; et al. Comparative Analysis of Viral RNA Signatures on Different RIG-I-like Receptors. Elife 2016, 5, e11275. [Google Scholar] [CrossRef] [PubMed]
- Eng, H.L.; Hsu, Y.Y.; Lin, T.M. Differences in TLR7/8 Activation between Monocytes and Macrophages. Biochem. Biophys. Res. Commun. 2018, 497, 319–325. [Google Scholar] [CrossRef]
- Neighbours, L.M.; Long, K.; Whitmore, A.C.; Heise, M.T. Myd88-Dependent Toll-Like Receptor 7 Signaling Mediates Protection from Severe Ross River Virus-Induced Disease in Mice. J. Virol. 2012, 86, 10675–10685. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, S.; Koarai, A.; Sugiura, H.; Ichikawa, T.; Kanda, M.; Tanaka, R.; Akamatsu, K.; Hirano, T.; Matsunaga, K.; Minakata, Y.; et al. Oxidative Stress Augments Toll-like Receptor 8 Mediated Neutrophilic Responses in Healthy Subjects. Respir. Res. 2009, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.S.; Shrihari, S.; Hykes, B.L.; Handley, S.A.; Andhey, P.S.; Huang, Y.J.S.; Swain, A.; Droit, L.; Chebrolu, K.K.; Mack, M.; et al. The Intestinal Microbiome Restricts Alphavirus Infection and Dissemination through a Bile Acid-Type I IFN Signaling Axis. Cell 2020, 182, 901–918.e18. [Google Scholar] [CrossRef]
- Bencze, D.; Fekete, T.; Pázmándi, K. Type i Interferon Production of Plasmacytoid Dendritic Cells under Control. Int. J. Mol. Sci. 2021, 22, 4190. [Google Scholar]
- Mapalagamage, M.; Weiskopf, D.; Sette, A.; De Silva, A.D. Current Understanding of the Role of T Cells in Chikungunya, Dengue and Zika Infections. Viruses 2022, 14, 242. [Google Scholar]
- Seymour, R.L.; Adams, A.P.; Leal, G.; Alcorn, M.D.H.; Weaver, S.C. A Rodent Model of Chikungunya Virus Infection in RAG1 -/- Mice, with Features of Persistence, for Vaccine Safety Evaluation. PLoS Negl. Trop. Dis. 2015, 9, e0003800. [Google Scholar] [CrossRef]
- Poh, C.M.; Chan, Y.H.; Ng, L.F.P. Role of T Cells in Chikungunya Virus Infection and Utilizing Their Potential in Anti-Viral Immunity. Front. Immunol. 2020, 11, 287. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex Differences in Immune Responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar]
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Solanich, X.; Vargas-Parra, G.; van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; Antolí, A.; del Valle, J.; Rocamora-Blanch, G.; Setién, F.; Esteller, M.; et al. Genetic Screening for TLR7 Variants in Young and Previously Healthy Men with Severe COVID-19. Front. Immunol. 2021, 12, 2965. [Google Scholar] [CrossRef]
- Tramontana, F.; Battisti, S.; Napoli, N.; Strollo, R. Immuno-Endocrinology of COVID-19: The Key Role of Sex Hormones. Front. Endocrinol. 2021, 12, 726696. [Google Scholar] [CrossRef]
- Rettew, J.A.; Huet-Hudson, Y.M.; Marriott, I. Testosterone Reduces Macrophage Expression in the Mouse of Toll-like Receptor 4, a Trigger for Inflammation and Innate Immunity. Biol. Reprod. 2008, 78, 432–437. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, P.; Milano, S.; Barbera, C.; Di Bella, G.; La Rosa, M.; Ferlazzo, V.; Farrugio, R.; Miceli, D.M.; Miele, M.; Castagnetta, L.; et al. Sex Hormones Modulate Inflammatory Mediators Produced by Macrophages. Ann. N. Y Acad. Sci. 2006, 876, 426–429. [Google Scholar] [CrossRef]
- Liva, S.M.; Voskuhl, R.R. Testosterone Acts Directly on CD4 + T Lymphocytes to Increase IL-10 Production. J. Immunol. 2001, 167, 2060–2067. [Google Scholar] [CrossRef]
- Bobjer, J.; Katrinaki, M.; Tsatsanis, C.; Lundberg Giwercman, Y.; Giwercman, A. Negative Association between Testosterone Concentration and Inflammatory Markers in Young Men: A Nested Cross-Sectional Study. PLoS ONE 2013, 8, e61466. [Google Scholar] [CrossRef]
- Kalinchenko, S.Y.; Tishova, Y.A.; Mskhalaya, G.J.; Gooren, L.J.G.; Giltay, E.J.; Saad, F. Effects of Testosterone Supplementation on Markers of the Metabolic Syndrome and Inflammation in Hypogonadal Men with the Metabolic Syndrome: The Double-Blinded Placebo-Controlled Moscow Study. Clin. Endocrinol. 2010, 73, 602–612. [Google Scholar] [CrossRef]
- Lin, A.A.; Wojciechowski, S.E.; Hildeman, D.A. Androgens Suppress Antigen-Specific T Cell Responses and IFN-γ Production during Intracranial LCMV Infection. J. Neuroimmunol. 2010, 226, 8–19. [Google Scholar] [CrossRef]
- Rettew, J.A.; Huet, Y.M.; Marriott, I. Estrogens Augment Cell Surface TLR4 Expression on Murine Macrophages and Regulate Sepsis Susceptibility in Vivo. Endocrinology 2009, 150, 3877–3884. [Google Scholar] [CrossRef]
- Bouman, A.; Jan Heineman, M.; Faas, M.M. Sex Hormones and the Immune Response in Humans. Hum. Reprod. Update 2005, 11, 411–423. [Google Scholar]
- Seillet, C.; Laffont, S.; Tré, F.; Rouquié, N.; Ribot, C.; Arnal, J.-F.; Douin-Echinard, V.; Gourdy, P.; Guéry, J.-C. The TLR-Mediated Response of Plasmacytoid Dendritic Cells Is Positively Regulated by Estradiol in Vivo through Cell-Intrinsic Estrogen Receptor Signaling. Blood J. Am. Soc. Hematol. 2012, 119, 454–464. [Google Scholar] [CrossRef]
- Paharkova-Vatchkova, V.; Maldonado, R.; Kovats, S. Estrogen Preferentially Promotes the Differentiation of CD11c + CD11b Intermediate Dendritic Cells from Bone Marrow Precursors. J. Immunol. 2004, 172, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Hagen, S.H.; Henseling, F.; Hennesen, J.; Savel, H.; Delahaye, S.; Richert, L.; Ziegler, S.M.; Altfeld, M. Heterogeneous Escape from X Chromosome Inactivation Results in Sex Differences in Type I IFN Responses at the Single Human PDC Level. Cell. Rep. 2020, 33, 1–10. [Google Scholar] [CrossRef]
- Jacobsen, H.; Klein, S.L. Sex Differences in Immunity to Viral Infections. Front. Immunol. 2021, 12, 720952. [Google Scholar]
- Hsu, C.H.; Cruz-Lopez, F.; Vargas Torres, D.; Perez-Padilla, J.; Lorenzi, O.D.; Rivera, A.; Staples, J.E.; Lugo, E.; Munoz-Jordan, J.; Fischer, M.; et al. Risk Factors for Hospitalization of Patients with Chikungunya Virus Infection at Sentinel Hospitals in Puerto Rico. PLoS Negl. Trop. Dis. 2019, 13, e0007084. [Google Scholar] [CrossRef]
- Constant, L.E.C.; Rajsfus, B.F.; Carneiro, P.H.; Sisnande, T.; Mohana-Borges, R.; Allonso, D. Overview on Chikungunya Virus Infection: From Epidemiology to State-of-the-Art Experimental Models. Front. Microbiol. 2021, 12, 744164. [Google Scholar]
- Barr, K.L.; Vaidhyanathan, V. Chikungunya in Infants and Children: Is Pathogenesis Increasing? Viruses 2019, 11, 294. [Google Scholar]
- Agrawal, A.; Agrawal, S.; Cao, J.-N.; Su, H.; Osann, K.; Gupta, S. Altered Innate Immune Functioning of Dendritic Cells in Elderly Humans: A Role of Phosphoinositide 3-Kinase-Signaling Pathway. J. Immunol. 2007, 178, 6912–6922. [Google Scholar] [CrossRef]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-Dependent Dysregulation of Innate Immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar]
- Tseng, C.W.; Kyme, P.A.; Arruda, A.; Ramanujan, V.K.; Tawackoli, W.; Liu, G.Y. Innate Immune Dysfunctions in Aged Mice Facilitate the Systemic Dissemination of Methicillin-Resistant S. Aureus. PLoS ONE 2012, 7, e41454. [Google Scholar] [CrossRef]
- Nogusa, S.; Ritz, B.W.; Kassim, S.H.; Jennings, S.R.; Gardner, E.M. Characterization of Age-Related Changes in Natural Killer Cells during Primary Influenza Infection in Mice. Mech. Ageing Dev. 2008, 129, 223–230. [Google Scholar] [CrossRef]
- Beli, E.; Clinthorne, J.F.; Duriancik, D.M.; Hwang, I.; Kim, S.; Gardner, E.M. Natural Killer Cell Function Is Altered during the Primary Response of Aged Mice to Influenza Infection. Mech. Ageing Dev. 2011, 132, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Kissin, E.; Tomasi, M.; McCartney-Francis, N.; Gibbs, C.L.; Smith, P.D. Age-Related Decline in Murine Macrophage Production of Nitric Oxide. J. Infect. Dis. 1997, 175, 1004–1007. [Google Scholar]
- Renshaw, M.; Rockwell, J.; Engleman, C.; Gewirtz, A.; Katz, J.; Sambhara, S. Cutting Edge: Impaired Toll-Like Receptor Expression and Function in Aging. J. Immunol. 2002, 169, 4697–4701. [Google Scholar] [CrossRef] [PubMed]
- Moretto, M.M.; Lawlor, E.M.; Khan, I.A. Aging Mice Exhibit a Functional Defect in Mucosal Dendritic Cell Response against an Intracellular Pathogen. J. Immunol. 2008, 181, 7977–7984. [Google Scholar] [CrossRef]
- Fong, S.-W.; Kini, R.M.; Ng, L.F.P. Mosquito Saliva Reshapes Alphavirus Infection and Immunopathogenesis. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef]
- Agarwal, A.; Joshi, G.; Nagar, D.P.; Sharma, A.K.; Sukumaran, D.; Pant, S.C.; Parida, M.M.; Dash, P.K. Mosquito Saliva Induced Cutaneous Events Augment Chikungunya Virus Replication and Disease Progression. Infect. Genet. Evol. 2016, 40, 126–135. [Google Scholar] [CrossRef]
- Surasombatpattana, P.; Ekchariyawat, P.; Hamel, R.; Patramool, S.; Thongrungkiat, S.; Denizot, M.; Delaunay, P.; Thomas, F.; Luplertlop, N.; Yssel, H.; et al. Aedes Aegypti Saliva Contains a Prominent 34-KDa Protein That Strongly Enhances Dengue Virus Replication in Human Keratinocytes. J. Investig. Dermatol. 2014, 134, 281–284. [Google Scholar] [CrossRef]
- Pingen, M.; Schmid, M.A.; Harris, E.; McKimmie, C.S. Mosquito Biting Modulates Skin Response to Virus Infection. Trends Parasitol. 2017, 33, 645–657. [Google Scholar]
- Pingen, M.; Bryden, S.R.; Pondeville, E.; Schnettler, E.; Kohl, A.; Merits, A.; Fazakerley, J.K.; Graham, G.J.; McKimmie, C.S. Host Inflammatory Response to Mosquito Bites Enhances the Severity of Arbovirus Infection. Immunity 2016, 44, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Thangamani, S.; Higgs, S.; Ziegler, S.; Vanlandingham, D.; Tesh, R.; Wikel, S. Host Immune Response to Mosquito-Transmitted Chikungunya Virus Differs from That Elicited by Needle Inoculated Virus. PLoS ONE 2010, 5, e12137. [Google Scholar] [CrossRef] [PubMed]
- Wichit, S.; Diop, F.; Hamel, R.; Talignani, L.; Ferraris, P.; Cornelie, S.; Liegeois, F.; Thomas, F.; Yssel, H.; Missé, D. Aedes Aegypti Saliva Enhances Chikungunya Virus Replication in Human Skin Fibroblasts via Inhibition of the Type I Interferon Signaling Pathway. Infect. Genet. Evol. 2017, 55, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Hibl, B.M.; Dailey Garnes, N.J.M.; Kneubehl, A.R.; Vogt, M.B.; Spencer Clinton, J.L.; Rico-Hesse, R.R. Mosquito-Bite Infection of Humanized Mice with Chikungunya Virus Produces Systemic Disease with Long-Term Effects. PLoS Negl. Trop. Dis. 2021, 15, e0009427. [Google Scholar] [CrossRef]
- Surasombatpattana, P.; Hamel, R.; Patramool, S.; Luplertlop, N.; Thomas, F.; Desprès, P.; Briant, L.; Yssel, H.; Missé, D. Dengue Virus Replication in Infected Human Keratinocytes Leads to Activation of Antiviral Innate Immune Responses. Infect. Genet. Evol. 2011, 11, 1664–1673. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, M.; Rayner, J.O. Immune Response to Chikungunya Virus: Sex as a Biological Variable and Implications for Natural Delivery via the Mosquito. Viruses 2023, 15, 1869. https://doi.org/10.3390/v15091869
Taylor M, Rayner JO. Immune Response to Chikungunya Virus: Sex as a Biological Variable and Implications for Natural Delivery via the Mosquito. Viruses. 2023; 15(9):1869. https://doi.org/10.3390/v15091869
Chicago/Turabian StyleTaylor, Meagan, and Jonathan O. Rayner. 2023. "Immune Response to Chikungunya Virus: Sex as a Biological Variable and Implications for Natural Delivery via the Mosquito" Viruses 15, no. 9: 1869. https://doi.org/10.3390/v15091869
APA StyleTaylor, M., & Rayner, J. O. (2023). Immune Response to Chikungunya Virus: Sex as a Biological Variable and Implications for Natural Delivery via the Mosquito. Viruses, 15(9), 1869. https://doi.org/10.3390/v15091869