

Airway Epithelial-Derived Immune Mediators in COVID-19

Abstract
1. Introduction
2. Main
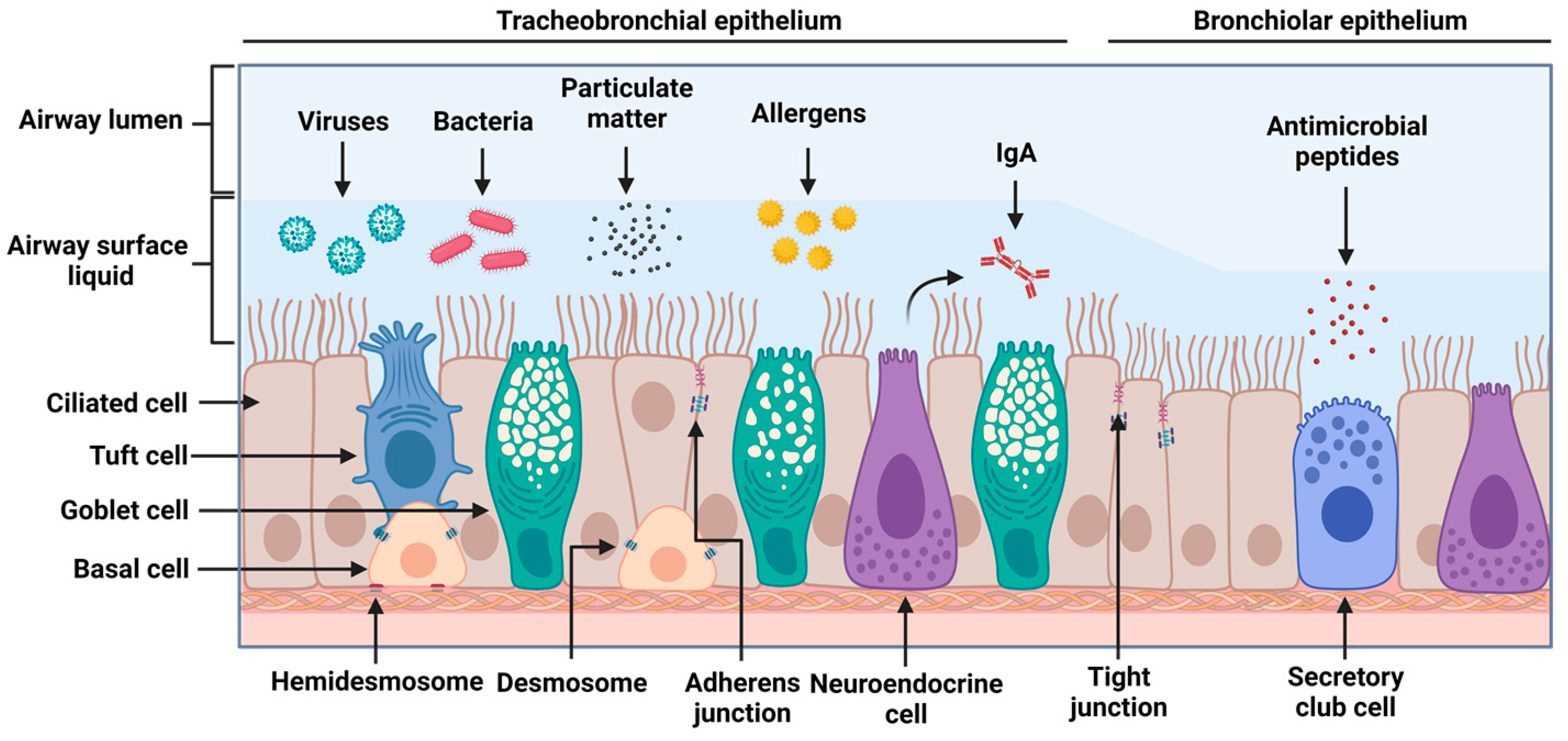
2.1. Airway Epithelial Structure and Function
2.2. Epithelial Machinery and Secreted Factors in Viral Infections
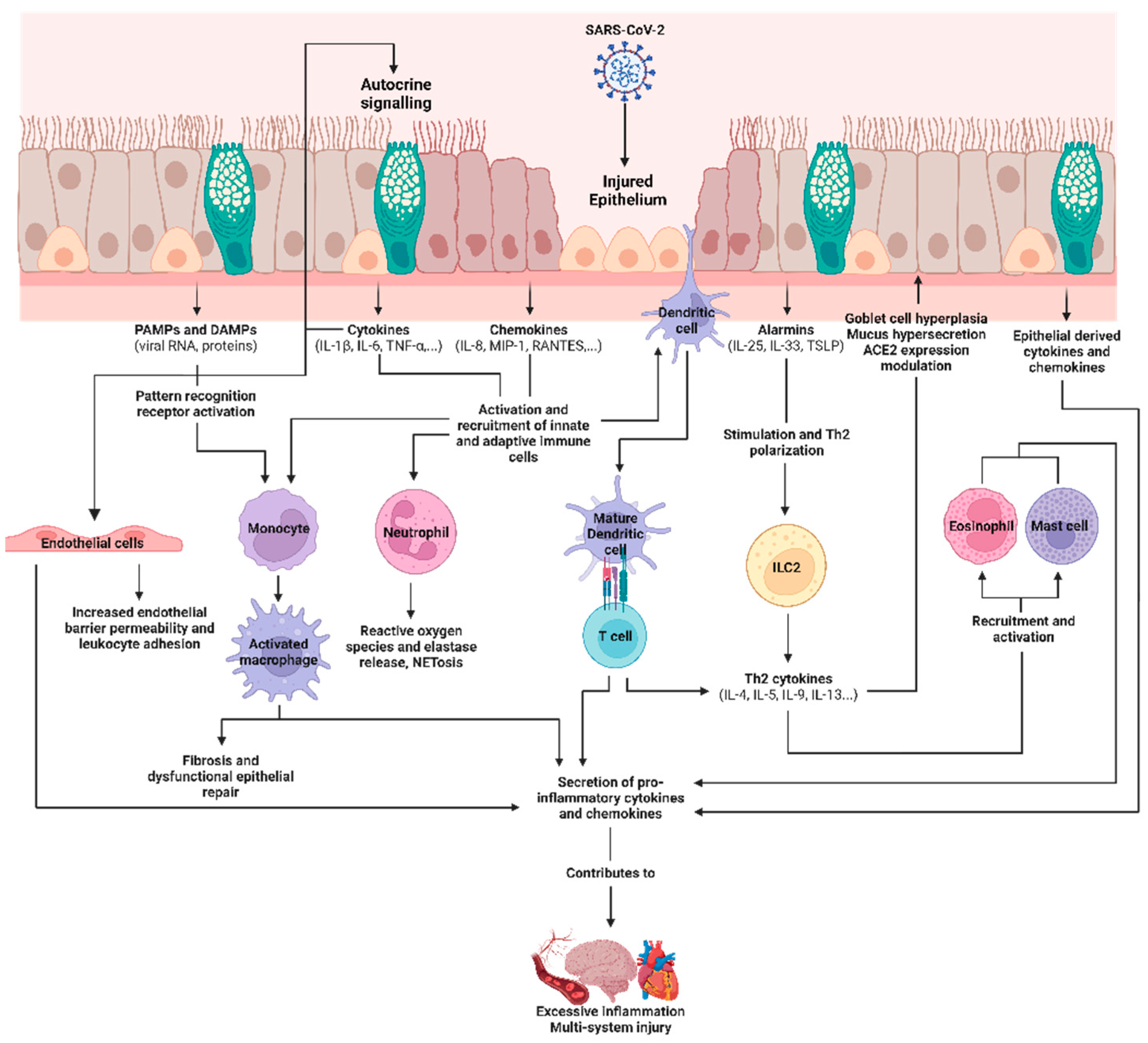
2.3. COVID-19, SARS-CoV-2, and the Airway Epithelium
2.4. Epithelial-Derived Immune Mediators in SARS-CoV-2 Infections and Their Roles as Biomarkers

{kind=link}
{kind=link}
{kind=link}
| Immune Mediators Upregulated or Secreted in the Airway Epithelium in Response to SARS-CoV-2 Infection and COVID-19 | Role in COVID-19 |
|---|---|
| Chemokines CXCL1, CXCL3, CXCL6, CXCL17 [87] IL-8 [89,142] CXCL2 [143] |
|
| Pro-inflammatory cytokines IL-1β [89,146] IL-10 [146,147] IL-6 [89,105,142] TNF-α [89,142] | |
| Epithelial-derived alarmins IL-33 [111] TSLP [123] |
2.5. Current and Future Strategies for COVID-19 Therapy
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clinical Spectrum. Available online: https://www.covid19treatmentguidelines.nih.gov/overview/clinical-spectrum/ (accessed on 27 March 2023).
- Ofner, M.; Salvadori, M.; Chung, Y.-E.; Green, M.; Pucchio, A.; Gravel-Tropper, D.; Plamodon, M.; Rajda, E. COVID-19 Signs, Symptoms and Severity of Disease: A Clinician Guide. Available online: https://www.canada.ca/en/public-health/services/diseases/2019-novel-coronavirus-infection/guidance-documents/signs-symptoms-severity.html (accessed on 15 May 2023).
- Shanmugam, C.; Mohammed, A.R.; Ravuri, S.; Luthra, V.; Rajagopal, N.; Karre, S. COVID-2019—A Comprehensive Pathology Insight. Pathol. Res. Pract. 2020, 216, 153222. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Imai, R.; Tomishima, Y.; Nishimura, N. Progressive Pulmonary Fibrosis Due to Diffuse Alveolar Damage in a COVID-19-infected Autopsy Case. Respirol. Case Rep. 2022, 10, e0934. [Google Scholar] [CrossRef] [PubMed]
- Zaim, S.; Chong, J.H.; Sankaranarayanan, V.; Harky, A. COVID-19 and Multiorgan Response. Curr. Probl. Cardiol. 2020, 45, 100618. [Google Scholar] [CrossRef]
- Hasan, S.S.; Capstick, T.; Ahmed, R.; Kow, C.S.; Mazhar, F.; Merchant, H.A.; Zaidi, S.T.R. Mortality in COVID-19 Patients with Acute Respiratory Distress Syndrome and Corticosteroids Use: A Systematic Review and Meta-Analysis. Expert Rev. Respir. Med. 2020, 14, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Rostami, M.R.; Leopold, P.L.; Mezey, J.G.; O’Beirne, S.L.; Strulovici-Barel, Y.; Crystal, R.G. Expression of the SARS-CoV-2 ACE2 Receptor in the Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 2020, 202, 219–229. [Google Scholar] [CrossRef]
- Hewitt, R.J.; Lloyd, C.M. Regulation of Immune Responses by the Airway Epithelial Cell Landscape. Nat. Rev. Immunol. 2021, 21, 347–362. [Google Scholar] [CrossRef]
- Tam, A.; Wadsworth, S.; Dorscheid, D.; Man, S.F.P.; Sin, D.D. The Airway Epithelium: More than Just a Structural Barrier. Ther. Adv. Respir. Dis. 2011, 5, 255–273. [Google Scholar] [CrossRef]
- Knight, D.A.; Holgate, S.T. The Airway Epithelium: Structural and Functional Properties in Health and Disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef]
- Deprez, M.; Zaragosi, L.-E.; Truchi, M.; Becavin, C.; Ruiz García, S.; Arguel, M.-J.; Plaisant, M.; Magnone, V.; Lebrigand, K.; Abelanet, S.; et al. A Single-Cell Atlas of the Human Healthy Airways. Am. J. Respir. Crit. Care Med. 2020, 202, 1636–1645. [Google Scholar] [CrossRef]
- Tizzano, M.; Gulbransen, B.D.; Vandenbeuch, A.; Clapp, T.R.; Herman, J.P.; Sibhatu, H.M.; Churchill, M.E.A.; Silver, W.L.; Kinnamon, S.C.; Finger, T.E. Nasal Chemosensory Cells Use Bitter Taste Signaling to Detect Irritants and Bacterial Signals. Proc. Natl. Acad. Sci. USA 2010, 107, 3210–3215. [Google Scholar] [CrossRef]
- Kotas, M.E.; Moore, C.M.; Gurrola, J.G.; Pletcher, S.D.; Goldberg, A.N.; Alvarez, R.; Yamato, S.; Bratcher, P.E.; Shaughnessy, C.A.; Zeitlin, P.L.; et al. IL-13–Programmed Airway Tuft Cells Produce PGE2, Which Promotes CFTR-Dependent Mucociliary Function. JCI Insight 2022, 7, e159832. [Google Scholar] [CrossRef] [PubMed]
- Ruysseveldt, E.; Martens, K.; Steelant, B. Airway Basal Cells, Protectors of Epithelial Walls in Health and Respiratory Diseases. Front. Allergy 2021, 2, 787128. [Google Scholar] [CrossRef] [PubMed]
- Iber, D.; Vetter, R. 3D Organisation of Cells in Pseudostratified Epithelia. Front. Phys. 2022, 10, 898160. [Google Scholar] [CrossRef]
- Iwasaki, A.; Foxman, E.F.; Molony, R.D. Early Local Immune Defenses in the Respiratory Tract. Nat. Rev. Immunol. 2017, 17, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, P.K. The Development of Large and Small Airways. Am. J. Respir. Crit. Care Med. 1998, 157, S174–S180. [Google Scholar] [CrossRef]
- Verkman, A.S.; Song, Y.; Thiagarajah, J.R. Role of Airway Surface Liquid and Submucosal Glands in Cystic Fibrosis Lung Disease. Am. J. Physiol. Cell Physiol. 2003, 284, C2–C15. [Google Scholar] [CrossRef]
- Crystal, R.G.; Randell, S.H.; Engelhardt, J.F.; Voynow, J.; Sunday, M.E. Airway Epithelial Cells. Proc. Am. Thorac. Soc. 2008, 5, 772–777. [Google Scholar] [CrossRef]
- Rackley, C.R.; Stripp, B.R. Building and Maintaining the Epithelium of the Lung. J. Clin. Investig. 2012, 122, 2724–2730. [Google Scholar] [CrossRef]
- Ganesan, S.; Comstock, A.T.; Sajjan, U.S. Barrier Function of Airway Tract Epithelium. Tissue Barriers 2013, 1, e24997. [Google Scholar] [CrossRef]
- Reynolds, S.D.; Malkinson, A.M. Clara Cell: Progenitor for the Bronchiolar Epithelium. Int. J. Biochem. Cell Biol. 2010, 42, 1–4. [Google Scholar] [CrossRef]
- Curran, D.R.; Cohn, L. Advances in Mucous Cell Metaplasia. Am. J. Respir. Cell Mol. Biol. 2010, 42, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, F.; Georas, S.N. Breaking Barriers. New Insights into Airway Epithelial Barrier Function in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2014, 50, 857–869. [Google Scholar] [CrossRef]
- Pohl, C.; Hermanns, M.I.; Uboldi, C.; Bock, M.; Fuchs, S.; Dei-Anang, J.; Mayer, E.; Kehe, K.; Kummer, W.; Kirkpatrick, C.J. Barrier Functions and Paracellular Integrity in Human Cell Culture Models of the Proximal Respiratory Unit. Eur. J. Pharm. Biopharm. 2009, 72, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A. Dendritic Cell-Airway Epithelial Cell Cross-Talk Changes with Age and Contributes to Chronic Lung Inflammatory Diseases in the Elderly. Int. J. Mol. Sci. 2017, 18, 1206. [Google Scholar] [CrossRef]
- Gras, D.; Martinez-Anton, A.; Bourdin, A.; Garulli, C.; de Senneville, L.; Vachier, I.; Vitte, J.; Chanez, P. Human Bronchial Epithelium Orchestrates Dendritic Cell Activation in Severe Asthma. Eur. Respir. J. 2017, 49, 1602399. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Ostrowski, L.E. Cilia and Mucociliary Clearance. Cold Spring Harb. Perspect. Biol. 2017, 9, a028241. [Google Scholar] [CrossRef] [PubMed]
- Houtmeyers, E.; Gosselink, R.; Gayan-Ramirez, G.; Decramer, M. Regulation of Mucociliary Clearance in Health and Disease. Eur. Respir. J. 1999, 13, 1177–1188. [Google Scholar] [CrossRef]
- Parker, D.; Prince, A. Innate Immunity in the Respiratory Epithelium. Am. J. Respir. Cell Mol. Biol. 2011, 45, 189–201. [Google Scholar] [CrossRef]
- Singh, P.K.; Jia, H.P.; Wiles, K.; Hesselberth, J.; Liu, L.; Conway, B.A.; Greenberg, E.P.; Valore, E.V.; Welsh, M.J.; Ganz, T.; et al. Production of Beta-Defensins by Human Airway Epithelia. Proc. Natl. Acad. Sci. USA 1998, 95, 14961–14966. [Google Scholar] [CrossRef]
- Harcourt, J.L.; McDonald, M.; Svoboda, P.; Pohl, J.; Tatti, K.; Haynes, L.M. Human Cathelicidin, LL-37, Inhibits Respiratory Syncytial Virus Infection in Polarized Airway Epithelial Cells. BMC Res. Notes 2016, 9, 11. [Google Scholar] [CrossRef]
- Hui, K.P.-Y.; Cheung, M.-C.; Lai, K.-L.; Ng, K.-C.; Ho, J.C.-W.; Peiris, M.; Nicholls, J.M.; Chan, M.C.-W. Role of Epithelial–Endothelial Cell Interaction in the Pathogenesis of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection. Clin. Infect. Dis. 2021, 74, 199–209. [Google Scholar] [CrossRef]
- Johansen, F.-E.; Kaetzel, C. Regulation of the Polymeric Immunoglobulin Receptor and IgA Transport: New Advances in Environmental Factors That Stimulate PIgR Expression and Its Role in Mucosal Immunity. Mucosal Immunol. 2011, 4, 598–602. [Google Scholar] [CrossRef]
- Sánchez Montalvo, A.; Gohy, S.; Rombaux, P.; Pilette, C.; Hox, V. The Role of IgA in Chronic Upper Airway Disease: Friend or Foe? Front. Allergy 2022, 3, 26. [Google Scholar] [CrossRef]
- Rohmann, K.; Tschernig, T.; Pabst, R.; Goldmann, T.; Drömann, D. Innate Immunity in the Human Lung: Pathogen Recognition and Lung Disease. Cell Tissue Res. 2011, 343, 167–174. [Google Scholar] [CrossRef]
- Nigar, S.; Shimosato, T. Cooperation of Oligodeoxynucleotides and Synthetic Molecules as Enhanced Immune Modulators. Front. Nutr. 2019, 6, 140. [Google Scholar] [CrossRef]
- Zhou, R.; Liu, L.; Wang, Y. Viral Proteins Recognized by Different TLRs. J. Med. Virol. 2021, 93, 6116–6123. [Google Scholar] [CrossRef]
- Lester, S.N.; Li, K. Toll-Like Receptors in Antiviral Innate Immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef]
- de Oliviera Nascimento, L.; Massari, P.; Wetzler, L. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Ullah, M.O.; Sweet, M.J.; Mansell, A.; Kellie, S.; Kobe, B. TRIF-Dependent TLR Signaling, Its Functions in Host Defense and Inflammation, and Its Potential as a Therapeutic Target. J. Leukoc. Biol. 2016, 100, 27–45. [Google Scholar] [CrossRef]
- Erb, A.; Zissler, U.M.; Oelsner, M.; Chaker, A.M.; Schmidt-Weber, C.B.; Jakwerth, C.A. Genome-Wide Gene Expression Analysis Reveals Unique Genes Signatures of Epithelial Reorganization in Primary Airway Epithelium Induced by Type-I, -II and -III Interferons. Biosensors 2022, 12, 929. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like Receptors: Their Regulation and Roles in RNA Sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Johnston, S.L.; Goldblatt, D.L.; Evans, S.E.; Tuvim, M.J.; Dickey, B.F. Airway Epithelial Innate Immunity. Front. Physiol. 2021, 12, 749077. [Google Scholar] [CrossRef]
- Divekar, R.; Kita, H. Recent Advances in Epithelium-Derived Cytokines (IL-33, IL-25 and TSLP) and Allergic Inflammation. Curr. Opin. Allergy Clin. Immunol. 2015, 15, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Duchesne, M.; Okoye, I.; Lacy, P. Epithelial Cell Alarmin Cytokines: Frontline Mediators of the Asthma Inflammatory Response. Front. Immunol. 2022, 13, 975914. [Google Scholar] [CrossRef]
- Roan, F.; Obata-Ninomiya, K.; Ziegler, S.F. Epithelial Cell–Derived Cytokines: More than Just Signaling the Alarm. J. Clin. Investig. 2019, 129, 1441–1451. [Google Scholar] [CrossRef]
- Patel, N.N.; Kohanski, M.A.; Maina, I.W.; Workman, A.D.; Herbert, D.R.; Cohen, N.A. Sentinels at the Wall: Epithelial-Derived Cytokines Serve as Triggers of Upper Airway Type-2 Inflammation. Int. Forum Allergy Rhinol. 2019, 9, 93–99. [Google Scholar] [CrossRef]
- Murdaca, G.; Paladin, F.; Tonacci, A.; Borro, M.; Greco, M.; Gerosa, A.; Isola, S.; Allegra, A.; Gangemi, S. Involvement of Il-33 in the Pathogenesis and Prognosis of Major Respiratory Viral Infections: Future Perspectives for Personalized Therapy. Biomedicines 2022, 10, 715. [Google Scholar] [CrossRef]
- Shlomovitz, I.; Erlich, Z.; Speir, M.; Zargarian, S.; Baram, N.; Engler, M.; Edry-Botzer, L.; Munitz, A.; Croker, B.A.; Gerlic, M. Necroptosis Directly Induces the Release of Full-Length Biologically Active IL-33 In Vitro and in an Inflammatory Disease Model. FEBS J. 2019, 286, 507–522. [Google Scholar] [CrossRef]
- Drake, L.Y.; Kita, H. IL-33: Biological Properties, Functions and Roles in Airway Disease. Immunol. Rev. 2017, 278, 173–184. [Google Scholar] [CrossRef]
- Wu, Y.; Lai, A.C.; Chi, P.; Thio, C.L.; Chen, W.; Tsai, C.; Lee, Y.L.; Lukacs, N.W.; Chang, Y. Pulmonary IL-33 Orchestrates Innate Immune Cells to Mediate Respiratory Syncytial Virus-evoked Airway Hyperreactivity and Eosinophilia. Allergy 2020, 75, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shao, Z.; Shangguan, G.; Bie, Q.; Zhang, B. Biological Properties and the Role of IL-25 in Disease Pathogenesis. J. Immunol. Res. 2018, 2018, 6519465. [Google Scholar] [CrossRef] [PubMed]
- Doran, E.; Cai, F.; Holweg, C.T.J.; Wong, K.; Brumm, J.; Arron, J.R. Interleukin-13 in Asthma and Other Eosinophilic Disorders. Front. Med. 2017, 4, 139. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Cheung, P.F.Y.; Ip, W.K.; Lam, C.W.K. Interleukin-25-Induced Chemokines and Interleukin-6 Release from Eosinophils Is Mediated by P38 Mitogen-Activated Protein Kinase, c-Jun N-Terminal Kinase, and Nuclear Factor-KappaB. Am. J. Respir. Cell Mol. Biol. 2005, 33, 186–194. [Google Scholar] [CrossRef]
- Hough, K.P.; Curtiss, M.L.; Blain, T.J.; Liu, R.-M.; Trevor, J.; Deshane, J.S.; Thannickal, V.J. Airway Remodeling in Asthma. Front. Med. (Lausanne) 2020, 7, 191. [Google Scholar] [CrossRef]
- Gregory, L.G.; Jones, C.P.; Walker, S.A.; Sawant, D.; Gowers, K.H.C.; Campbell, G.A.; McKenzie, A.N.J.; Lloyd, C.M. IL-25 Drives Remodelling in Allergic Airways Disease Induced by House Dust Mite. Thorax 2013, 68, 82–90. [Google Scholar] [CrossRef]
- Vareille, M.; Kieninger, E.; Edwards, M.R.; Regamey, N. The Airway Epithelium: Soldier in the Fight against Respiratory Viruses. Clin. Microbiol. Rev. 2011, 24, 210–229. [Google Scholar] [CrossRef]
- Patel, J.A.; Jiang, Z.; Nakajima, N.; Kunimoto, M. Autocrine Regulation of Interleukin-8 by Interleukin-1alpha in Respiratory Syncytial Virus-Infected Pulmonary Epithelial Cells In Vitro. Immunology 1998, 95, 501–506. [Google Scholar] [CrossRef]
- Glaser, L.; Coulter, P.J.; Shields, M.; Touzelet, O.; Power, U.F.; Broadbent, L. Airway Epithelial Derived Cytokines and Chemokines and Their Role in the Immune Response to Respiratory Syncytial Virus Infection. Pathogens 2019, 8, 106. [Google Scholar] [CrossRef]
- Calvert, B.; Quiroz, E.; Lorenzana, Z.; Doan, N.; Kim, S.; Senger, C.; Wallace, W.; Salomon, M.; Henley, J.; Ryan, A. Neutrophil-Epithelial Interactions Augment Infectivity and pro-Inflammatory Responses to SARS-CoV-2 Infection. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cavalcante-Silva, L.H.A.; Carvalho, D.C.M.; Lima, É.D.A.; Galvão, J.G.F.M.; Silva, J.S.D.F.D.; de Sales-Neto, J.M.; Rodrigues-Mascarenhas, S. Neutrophils and COVID-19: The Road so Far. Int. Immunopharmacol. 2021, 90, 107233. [Google Scholar] [CrossRef]
- Schönrich, G.; Raftery, M.J. Neutrophil Extracellular Traps Go Viral. Front. Immunol. 2016, 7, 366. [Google Scholar] [CrossRef]
- Puttur, F.; Gregory, L.G.; Lloyd, C.M. Airway Macrophages as the Guardians of Tissue Repair in the Lung. Immunol. Cell Biol. 2019, 97, 246–257. [Google Scholar] [CrossRef]
- Barreto-Duran, E.; Szczepański, A.; Gałuszka-Bulaga, A.; Surmiak, M.; Siedlar, M.; Sanak, M.; Rajfur, Z.; Milewska, A.; Lenart, M.; Pyrć, K. The Interplay between the Airway Epithelium and Tissue Macrophages during the SARS-CoV-2 Infection. Front. Immunol. 2022, 13, 991991. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Trougakos, I.P.; Stamatelopoulos, K.; Terpos, E.; Tsitsilonis, O.E.; Aivalioti, E.; Paraskevis, D.; Kastritis, E.; Pavlakis, G.N.; Dimopoulos, M.A. Insights to SARS-CoV-2 Life Cycle, Pathophysiology, and Rationalized Treatments That Target COVID-19 Clinical Complications. J. Biomed. Sci. 2021, 28, 9. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Lee, I.T.; Nakayama, T.; Wu, C.-T.; Goltsev, Y.; Jiang, S.; Gall, P.A.; Liao, C.-K.; Shih, L.-C.; Schürch, C.M.; McIlwain, D.R.; et al. ACE2 Localizes to the Respiratory Cilia and Is Not Increased by ACE Inhibitors or ARBs. Nat. Commun. 2020, 11, 5453. [Google Scholar] [CrossRef]
- Roddy, J.T.; Benn, B.S.; Lwin, P.E.; Kutty, R.; Yelisetty, A.; Darisetty, S.; Kurman, J.S. Diagnosis of COVID-19 from Lower Airway Sampling after Negative Nasopharyngeal Swab. J. Public Health Emerg. 2021, 5. [Google Scholar] [CrossRef]
- Wölfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological Assessment of Hospitalized Patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef]
- Schaefer, I.-M.; Padera, R.F.; Solomon, I.H.; Kanjilal, S.; Hammer, M.M.; Hornick, J.L.; Sholl, L.M. In Situ Detection of SARS-CoV-2 in Lungs and Airways of Patients with COVID-19. Mod. Pathol. 2020, 33, 2104–2114. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.-Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.-X.; Gong, L.; et al. CD147-Spike Protein Is a Novel Route for SARS-CoV-2 Infection to Host Cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Felgenhauer, U.; Schoen, A.; Gad, H.H.; Hartmann, R.; Schaubmar, A.R.; Failing, K.; Drosten, C.; Weber, F. Inhibition of SARS–CoV-2 by Type I and Type III Interferons. J. Biol. Chem. 2020, 295, 13958–13964. [Google Scholar] [CrossRef] [PubMed]
- Wark, P.A.B.; Pathinayake, P.S.; Kaiko, G.; Nichol, K.; Ali, A.; Chen, L.; Sutanto, E.N.; Garratt, L.W.; Sohal, S.S.; Lu, W.; et al. ACE2 Expression Is Elevated in Airway Epithelial Cells from Older and Male Healthy Individuals but Reduced in Asthma. Respirology 2021, 26, 442–451. [Google Scholar] [CrossRef]
- Knight, S.C.; McCurdy, S.R.; Rhead, B.; Coignet, M.V.; Park, D.S.; Roberts, G.H.L.; Berkowitz, N.D.; Zhang, M.; Turissini, D.; Delgado, K.; et al. COVID-19 Susceptibility and Severity Risks in a Cross-Sectional Survey of over 500,000 US Adults. BMJ Open 2022, 12, e049657. [Google Scholar] [CrossRef] [PubMed]
- Mulay, A.; Konda, B.; Garcia, G.; Yao, C.; Beil, S.; Villalba, J.M.; Koziol, C.; Sen, C.; Purkayastha, A.; Kolls, J.K.; et al. SARS-CoV-2 Infection of Primary Human Lung Epithelium for COVID-19 Modeling and Drug Discovery. Cell Rep. 2021, 35, 109055. [Google Scholar] [CrossRef]
- Milewska, A.; Kula-Pacurar, A.; Wadas, J.; Suder, A.; Szczepanski, A.; Dabrowska, A.; Owczarek, K.; Marcello, A.; Ochman, M.; Stacel, T.; et al. Replication of Severe Acute Respiratory Syndrome Coronavirus 2 in Human Respiratory Epithelium. J. Virol. 2020, 94, e00957-20. [Google Scholar] [CrossRef]
- Hao, S.; Ning, K.; Kuz, C.A.; Vorhies, K.; Yan, Z.; Qiu, J. Long-Term Modeling of SARS-CoV-2 Infection of In Vitro Cultured Polarized Human Airway Epithelium. mBio 2020, 11, e02852-20. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Do, T.N.D.; Claes, S.; Schols, D.; Neyts, J.; Jochmans, D. SARS-CoV-2 Virion Infectivity and Cytokine Production in Primary Human Airway Epithelial Cells. Viruses 2022, 14, 951. [Google Scholar] [CrossRef]
- Kasper, B.; Yue, X.; Goldmann, T.; Gülsen, A.; Kugler, C.; Yu, X.; Petersen, F. Air Exposure and Cell Differentiation Are Essential for Investigation of SARS-CoV-2 Entry Genes in Human Primary Airway Epithelial Cells in Vitro. Front. Med. 2022, 9, 897695. [Google Scholar] [CrossRef]
- Zhu, N.; Wang, W.; Liu, Z.; Liang, C.; Wang, W.; Ye, F.; Huang, B.; Zhao, L.; Wang, H.; Zhou, W.; et al. Morphogenesis and Cytopathic Effect of SARS-CoV-2 Infection in Human Airway Epithelial Cells. Nat. Commun. 2020, 11, 3910. [Google Scholar] [CrossRef]
- Bissonnette, E.Y.; Lauzon-Joset, J.-F.; Debley, J.S.; Ziegler, S.F. Cross-Talk Between Alveolar Macrophages and Lung Epithelial Cells Is Essential to Maintain Lung Homeostasis. Front. Immunol. 2020, 11, 583042. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 Severity Correlates with Airway Epithelium–Immune Cell Interactions Identified by Single-Cell Analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Thorne, L.G.; Reuschl, A.; Zuliani-Alvarez, L.; Whelan, M.V.X.; Turner, J.; Noursadeghi, M.; Jolly, C.; Towers, G.J. SARS-CoV-2 Sensing by RIG-I and MDA5 Links Epithelial Infection to Macrophage Inflammation. EMBO J. 2021, 40, e107826. [Google Scholar] [CrossRef]
- Deinhardt-Emmer, S.; Böttcher, S.; Häring, C.; Giebeler, L.; Henke, A.; Zell, R.; Jungwirth, J.; Jordan, P.M.; Werz, O.; Hornung, F.; et al. SARS-CoV-2 Causes Severe Epithelial Inflammation and Barrier Dysfunction. J. Virol. 2021, 95, e00110-21. [Google Scholar] [CrossRef]
- Biering, S.B.; Gomes de Sousa, F.T.; Tjang, L.V.; Pahmeier, F.; Zhu, C.; Ruan, R.; Blanc, S.F.; Patel, T.S.; Worthington, C.M.; Glasner, D.R.; et al. SARS-CoV-2 Spike Triggers Barrier Dysfunction and Vascular Leak via Integrins and TGF-β Signaling. Nat. Commun. 2022, 13, 7630. [Google Scholar] [CrossRef]
- Jover, E.; Matilla, L.; Garaikoetxea, M.; Fernández-Celis, A.; Muntendam, P.; Jaisser, F.; Rossignol, P.; López-Andrés, N. Beneficial Effects of Mineralocorticoid Receptor Pathway Blockade against Endothelial Inflammation Induced by SARS-CoV-2 Spike Protein. Biomedicines 2021, 9, 639. [Google Scholar] [CrossRef]
- Raghavan, S.; Kenchappa, D.B.; Leo, M.D. SARS-CoV-2 Spike Protein Induces Degradation of Junctional Proteins That Maintain Endothelial Barrier Integrity. Front. Cardiovasc. Med. 2021, 8, 582. [Google Scholar] [CrossRef]
- McCracken, I.R.; Saginc, G.; He, L.; Huseynov, A.; Daniels, A.; Fletcher, S.; Peghaire, C.; Kalna, V.; Andaloussi-Mäe, M.; Muhl, L.; et al. Lack of Evidence of Angiotensin-Converting Enzyme 2 Expression and Replicative Infection by SARS-CoV-2 in Human Endothelial Cells. Circulation 2021, 143, 865–868. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Weng, J. Endothelial Dysfunction in COVID-19: An Overview of Evidence, Biomarkers, Mechanisms and Potential Therapies. Acta Pharmacol. Sin. 2023, 44, 695–709. [Google Scholar] [CrossRef]
- Bortolotti, D.; Gentili, V.; Rizzo, S.; Schiuma, G.; Beltrami, S.; Strazzabosco, G.; Fernandez, M.; Caccuri, F.; Caruso, A.; Rizzo, R. TLR3 and TLR7 RNA Sensor Activation during SARS-CoV-2 Infection. Microorganisms 2021, 9, 1820. [Google Scholar] [CrossRef]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 Spike Protein Induces Inflammation via TLR2-Dependent Activation of the NF-ΚB Pathway. eLife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Contoli, M.; Papi, A.; Tomassetti, L.; Rizzo, P.; Vieceli Dalla Sega, F.; Fortini, F.; Torsani, F.; Morandi, L.; Ronzoni, L.; Zucchetti, O.; et al. Blood Interferon-α Levels and Severity, Outcomes, and Inflammatory Profiles in Hospitalized COVID-19 Patients. Front. Immunol. 2021, 12, 648004. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Kouwaki, T.; Nishimura, T.; Wang, G.; Oshiumi, H. RIG-I-Like Receptor-Mediated Recognition of Viral Genomic RNA of Severe Acute Respiratory Syndrome Coronavirus-2 and Viral Escape From the Host Innate Immune Responses. Front. Immunol. 2021, 12, 700926. [Google Scholar] [CrossRef]
- Miorin, L.; Kehrer, T.; Sanchez-Aparicio, M.T.; Zhang, K.; Cohen, P.; Patel, R.S.; Cupic, A.; Makio, T.; Mei, M.; Moreno, E.; et al. SARS-CoV-2 Orf6 Hijacks Nup98 to Block STAT Nuclear Import and Antagonize Interferon Signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 28344–28354. [Google Scholar] [CrossRef]
- Lee, J.-H.; Koepke, L.; Kirchhoff, F.; Sparrer, K.M.J. Interferon Antagonists Encoded by SARS-CoV-2 at a Glance. Med. Microbiol. Immunol. 2022, 212, 125–131. [Google Scholar] [CrossRef]
- da Silva, R.P.; Gonçalves, J.I.B.; Zanin, R.F.; Schuch, F.B.; de Souza, A.P.D. Circulating Type I Interferon Levels and COVID-19 Severity: A Systematic Review and Meta-Analysis. Front. Immunol. 2021, 12, 657363. [Google Scholar] [CrossRef]
- Ravindra, N.G.; Alfajaro, M.M.; Gasque, V.; Huston, N.C.; Wan, H.; Szigeti-Buck, K.; Yasumoto, Y.; Greaney, A.M.; Habet, V.; Chow, R.D.; et al. Single-Cell Longitudinal Analysis of SARS-CoV-2 Infection in Human Airway Epithelium Identifies Target Cells, Alterations in Gene Expression, and Cell State Changes. PLoS Biol. 2021, 19, e3001143. [Google Scholar] [CrossRef]
- Hsu, R.-J.; Yu, W.-C.; Peng, G.-R.; Ye, C.-H.; Hu, S.; Chong, P.C.T.; Yap, K.Y.; Lee, J.Y.C.; Lin, W.-C.; Yu, S.-H. The Role of Cytokines and Chemokines in Severe Acute Respiratory Syndrome Coronavirus 2 Infections. Front. Immunol. 2022, 13, 832394. [Google Scholar] [CrossRef]
- Barnett, K.C.; Xie, Y.; Asakura, T.; Song, D.; Liang, K.; Taft-Benz, S.A.; Guo, H.; Yang, S.; Okuda, K.; Gilmore, R.C.; et al. An Epithelial-Immune Circuit Amplifies Inflammasome and IL-6 Responses to SARS-CoV-2. Cell Host Microbe 2023, 31, 243–259.e6. [Google Scholar] [CrossRef]
- Murakami, M.; Kamimura, D.; Hirano, T. Pleiotropy and Specificity: Insights from the Interleukin 6 Family of Cytokines. Immunity 2019, 50, 812–831. [Google Scholar] [CrossRef]
- Battagello, D.S.; Dragunas, G.; Klein, M.O.; Ayub, A.L.P.; Velloso, F.J.; Correa, R.G. Unpuzzling COVID-19: Tissue-Related Signaling Pathways Associated with SARS-CoV-2 Infection and Transmission. Clin. Sci. 2020, 134, 2137–2160. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Yang, Y.; Ma, H.; Li, Z.; Zhang, J.; Cheng, J.; Zhang, X.; Zhao, Y.; Xia, Z.; et al. The Role of Interleukin-6 in Monitoring Severe Case of Coronavirus Disease 2019. EMBO Mol. Med. 2020, 12, e12421. [Google Scholar] [CrossRef]
- Santa Cruz, A.; Mendes-Frias, A.; Oliveira, A.I.; Dias, L.; Matos, A.R.; Carvalho, A.; Capela, C.; Pedrosa, J.; Castro, A.G.; Silvestre, R. Interleukin-6 Is a Biomarker for the Development of Fatal Severe Acute Respiratory Syndrome Coronavirus 2 Pneumonia. Front. Immunol. 2021, 12, 613422. [Google Scholar] [CrossRef]
- Sebbar, E.; Choukri, M. Interleukin 6: A Biomarker for COVID-19 Progression. Mater. Today Proc. 2023, 72, 3351–3355. [Google Scholar] [CrossRef]
- Liang, Y.; Ge, Y.; Sun, J. IL-33 in COVID-19: Friend or Foe? Cell Mol. Immunol. 2021, 18, 1602–1604. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic Characteristics of Bronchoalveolar Lavage Fluid and Peripheral Blood Mononuclear Cells in COVID-19 Patients. Emerg. Microbes Infect. 2020, 9, 761. [Google Scholar] [CrossRef]
- Gaurav, R.; Anderson, D.R.; Radio, S.J.; Bailey, K.L.; England, B.R.; Mikuls, T.R.; Thiele, G.M.; Strah, H.M.; Romberger, D.J.; Wyatt, T.A.; et al. IL-33 Depletion in COVID-19 Lungs. Chest 2021, 160, 1656–1659. [Google Scholar] [CrossRef]
- Byers, D.E.; Alexander-Brett, J.; Patel, A.C.; Agapov, E.; Dang-Vu, G.; Jin, X.; Wu, K.; You, Y.; Alevy, Y.; Girard, J.-P.; et al. Long-Term IL-33–Producing Epithelial Progenitor Cells in Chronic Obstructive Lung Disease. J. Clin. Investig. 2013, 123, 3967–3982. [Google Scholar] [CrossRef]
- Burke, H.; Freeman, A.; Cellura, D.C.; Stuart, B.L.; Brendish, N.J.; Poole, S.; Borca, F.; Phan, H.T.T.; Sheard, N.; Williams, S.; et al. Inflammatory Phenotyping Predicts Clinical Outcome in COVID-19. Respir. Res. 2020, 21, 245. [Google Scholar] [CrossRef]
- Zizzo, G.; Cohen, P.L. Imperfect Storm: Is Interleukin-33 the Achilles Heel of COVID-19? Lancet Rheumatol. 2020, 2, e779–e790. [Google Scholar] [CrossRef]
- Dreis, C.; Ottenlinger, F.M.; Putyrski, M.; Ernst, A.; Huhn, M.; Schmidt, K.G.; Pfeilschifter, J.M.; Radeke, H.H. Tissue Cytokine IL-33 Modulates the Cytotoxic CD8 T Lymphocyte Activity During Nutrient Deprivation by Regulation of Lineage-Specific Differentiation Programs. Front. Immunol. 2019, 10, 1698. [Google Scholar] [CrossRef]
- Komai-Koma, M.; Wang, E.; Kurowska-Stolarska, M.; Li, D.; McSharry, C.; Xu, D. Interleukin-33 Promoting Th1 Lymphocyte Differentiation Dependents on IL-12. Immunobiology 2016, 221, 412–417. [Google Scholar] [CrossRef]
- Faustino, L.D.; Griffith, J.W.; Rahimi, R.A.; Nepal, K.; Hamilos, D.L.; Cho, J.L.; Medoff, B.D.; Moon, J.J.; Vignali, D.A.A.; Luster, A.D. Interleukin-33 Activates Regulatory T Cells to Suppress Innate Γδ T Cell Responses in the Lung. Nat. Immunol. 2020, 21, 1371–1383. [Google Scholar] [CrossRef]
- von Massow, G.; Oh, S.; Lam, A.; Gustafsson, K. Gamma Delta T Cells and Their Involvement in COVID-19 Virus Infections. Front. Immunol. 2021, 12, 741218. [Google Scholar] [CrossRef]
- Schuijs, M.J.; Png, S.; Richard, A.C.; Tsyben, A.; Hamm, G.; Stockis, J.; Garcia, C.; Pinaud, S.; Nicholls, A.; Ros, X.R.; et al. ILC2-Driven Innate Immune Checkpoint Mechanism Antagonizes NK Cell Antimetastatic Function in the Lung. Nat. Immunol. 2020, 21, 998–1009. [Google Scholar] [CrossRef]
- Kato, A.; Favoreto, S.; Avila, P.C.; Schleimer, R.P. TLR3- and Th2 Cytokine-Dependent Production of Thymic Stromal Lymphopoietin in Human Airway Epithelial Cells. J. Immunol. 2007, 179, 1080–1087. [Google Scholar] [CrossRef]
- Gerla, L.; Moitra, S.; Pink, D.; Govindasamy, N.; Duchesne, M.; Reklow, E.; Hillaby, A.; May, A.; Lewis, J.D.; Melenka, L.; et al. SARS-CoV-2-Induced TSLP Is Associated with Duration of Hospital Stay in COVID-19 Patients. Viruses 2023, 15, 556. [Google Scholar] [CrossRef]
- Semlali, A.; Jacques, E.; Koussih, L.; Gounni, A.S.; Chakir, J. Thymic Stromal Lymphopoietin–Induced Human Asthmatic Airway Epithelial Cell Proliferation through an IL-13–Dependent Pathway. J. Allergy Clin. Immunol. 2010, 125, 844–850. [Google Scholar] [CrossRef]
- Shubin, N.J.; Clauson, M.; Niino, K.; Kasprzak, V.; Tsuha, A.; Guga, E.; Bhise, G.; Acharya, M.; Snyder, J.M.; Debley, J.S.; et al. Thymic Stromal Lymphopoietin Protects in a Model of Airway Damage and Inflammation via Regulation of Caspase-1 Activity and Apoptosis Inhibition. Mucosal Immunol. 2020, 13, 584–594. [Google Scholar] [CrossRef]
- Petersen, B.C.; Dolgachev, V.; Rasky, A.; Lukacs, N.W. IL-17E (IL-25) and IL-17RB Promote Respiratory Syncytial Virus-Induced Pulmonary Disease. J. Leukoc. Biol. 2014, 95, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.C.; Loo, S.-L.; Nichol, K.S.; Reid, A.T.; Veerati, P.C.; Esneau, C.; Wark, P.A.B.; Grainge, C.L.; Knight, D.A.; Vincent, T.; et al. IL-25 Blockade Augments Antiviral Immunity during Respiratory Virus Infection. Commun. Biol. 2022, 5, 415. [Google Scholar] [CrossRef]
- Ullah, R.; Khan, J.; Basharat, N.; Huo, D.; Ud Din, A.; Wang, G. Evaluation of Cardiac Biomarkers and Expression Analysis of IL-1, IL-6, IL-10, IL-17, and IL-25 among COVID-19 Patients from Pakistan. Viruses 2022, 14, 2149. [Google Scholar] [CrossRef]
- Neidleman, J.; Luo, X.; Frouard, J.; Xie, G.; Gill, G.; Stein, E.S.; McGregor, M.; Ma, T.; George, A.F.; Kosters, A.; et al. SARS-CoV-2-Specific T Cells Exhibit Phenotypic Features of Helper Function, Lack of Terminal Differentiation, and High Proliferation Potential. Cell Rep. Med. 2020, 1, 100081. [Google Scholar] [CrossRef]
- Roncati, L.; Nasillo, V.; Lusenti, B.; Riva, G. Signals of Th2 Immune Response from COVID-19 Patients Requiring Intensive Care. Ann. Hematol. 2020, 99, 1419–1420. [Google Scholar] [CrossRef]
- Gil-Etayo, F.J.; Suàrez-Fernández, P.; Cabrera-Marante, O.; Arroyo, D.; Garcinuño, S.; Naranjo, L.; Pleguezuelo, D.E.; Allende, L.M.; Mancebo, E.; Lalueza, A.; et al. T-Helper Cell Subset Response Is a Determining Factor in COVID-19 Progression. Front. Cell Infect. Microbiol. 2021, 11, 624483. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal Analyses Reveal Immunological Misfiring in Severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 888. [Google Scholar] [CrossRef]
- Moran, T.M.; Isobe, H.; Fernandez-Sesma, A.; Schulman, J.L. Interleukin-4 Causes Delayed Virus Clearance in Influenza Virus-Infected Mice. J. Virol. 1996, 70, 5230–5235. [Google Scholar] [CrossRef]
- Zlei, M.; Sidorov, I.A.; Joosten, S.A.; Heemskerk, M.H.M.; Myeni, S.K.; Pothast, C.R.; de Brouwer, C.S.; Boomaars-van der Zanden, A.L.; van Meijgaarden, K.E.; Morales, S.T.; et al. Immune Determinants of Viral Clearance in Hospitalised COVID-19 Patients: Reduced Circulating Naïve CD4+ T Cell Counts Correspond with Delayed Viral Clearance. Cells 2022, 11, 2743. [Google Scholar] [CrossRef]
- Gibellini, L.; De Biasi, S.; Meschiari, M.; Gozzi, L.; Paolini, A.; Borella, R.; Mattioli, M.; Lo Tartaro, D.; Fidanza, L.; Neroni, A.; et al. Plasma Cytokine Atlas Reveals the Importance of TH2 Polarization and Interferons in Predicting COVID-19 Severity and Survival. Front. Immunol. 2022, 13, 2743. [Google Scholar] [CrossRef]
- Carapito, R.; Li, R.; Helms, J.; Carapito, C.; Gujja, S.; Rolli, V.; Guimaraes, R.; Malagon-Lopez, J.; Spinnhirny, P.; Lederle, A.; et al. Identification of Driver Genes for Critical Forms of COVID-19 in a Deeply Phenotyped Young Patient Cohort. Sci. Transl. Med. 2021, 14, eabj7521. [Google Scholar] [CrossRef]
- Huaux, F.; Liu, T.; McGarry, B.; Ullenbruch, M.; Phan, S.H. Dual Roles of IL-4 in Lung Injury and Fibrosis. J. Immunol. 2003, 170, 2083–2092. [Google Scholar] [CrossRef]
- Marone, G.; Granata, F.; Pucino, V.; Pecoraro, A.; Heffler, E.; Loffredo, S.; Scadding, G.W.; Varricchi, G. The Intriguing Role of Interleukin 13 in the Pathophysiology of Asthma. Front. Pharmacol. 2019, 10, 1387. [Google Scholar] [CrossRef]
- de Paula, C.B.V.; de Azevedo, M.L.V.; Nagashima, S.; Martins, A.P.C.; Malaquias, M.A.S.; Miggiolaro, A.F.R.D.S.; Júnior, J.D.S.M.; Avelino, G.; Carmo, L.A.P.D.; Carstens, L.B.; et al. IL-4/IL-13 Remodeling Pathway of COVID-19 Lung Injury. Sci. Rep. 2020, 10, 18689. [Google Scholar] [CrossRef]
- Barton, L.M.; Duval, E.J.; Stroberg, E.; Ghosh, S.; Mukhopadhyay, S. COVID-19 Autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 2020, 153, 725–733. [Google Scholar] [CrossRef]
- Stölting, H.; Baillon, L.; Frise, R.; Bonner, K.; Hewitt, R.J.; Molyneaux, P.L.; Gore, M.L.; Barclay, W.S.; Saglani, S.; Lloyd, C.M. Distinct Airway Epithelial Immune Responses after Infection with SARS-CoV-2 Compared to H1N1. Mucosal Immunol. 2022, 15, 952–963. [Google Scholar] [CrossRef]
- Chen, H.; Liu, W.; Wang, Y.; Liu, D.; Zhao, L.; Yu, J. SARS-CoV-2 Activates Lung Epithelial Cell Proinflammatory Signaling and Leads to Immune Dysregulation in COVID-19 Patients. eBioMedicine 2021, 70, 103500. [Google Scholar] [CrossRef]
- Cui, L.; Fang, Z.; De Souza, C.M.; Lerbs, T.; Guan, Y.; Li, I.; Charu, V.; Chen, S.-Y.; Weissman, I.; Wernig, G. Innate Immune Cell Activation Causes Lung Fibrosis in a Humanized Model of Long COVID. Proc. Natl. Acad. Sci. USA 2023, 120, e2217199120. [Google Scholar] [CrossRef]
- Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Chemokines and Chemokine Receptors during COVID-19 Infection. Comput. Struct. Biotechnol. J. 2021, 19, 976–988. [Google Scholar] [CrossRef]
- Calvert, B.A.; Quiroz, E.J.; Lorenzana, Z.; Doan, N.; Kim, S.; Senger, C.N.; Anders, J.J.; Wallace, W.D.; Salomon, M.P.; Henley, J.; et al. Neutrophilic Inflammation Promotes SARS-CoV-2 Infectivity and Augments the Inflammatory Responses in Airway Epithelial Cells. Front. Immunol. 2023, 14, 1112870. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Cytokine Storm and Leukocyte Changes in Mild versus Severe SARS-CoV-2 Infection: Review of 3939 COVID-19 Patients in China and Emerging Pathogenesis and Therapy Concepts|Journal of Leukocyte Biology|Oxford Academic. Available online: https://academic.oup.com/jleukbio/article/108/1/17/6884549?login=false (accessed on 19 May 2023).
- Donlan, A.N.; Sutherland, T.E.; Marie, C.; Preissner, S.; Bradley, B.T.; Carpenter, R.M.; Sturek, J.M.; Ma, J.Z.; Moreau, G.B.; Donowitz, J.R.; et al. IL-13 Is a Driver of COVID-19 Severity. JCI Insight 2021, 6, e150107. [Google Scholar] [CrossRef]
- Morrison, C.B.; Edwards, C.E.; Shaffer, K.M.; Araba, K.C.; Wykoff, J.A.; Williams, D.R.; Asakura, T.; Dang, H.; Morton, L.C.; Gilmore, R.C.; et al. SARS-CoV-2 Infection of Airway Cells Causes Intense Viral and Cell Shedding, Two Spreading Mechanisms Affected by IL-13. Proc. Natl. Acad. Sci. USA 2022, 119, e2119680119. [Google Scholar] [CrossRef]
- Bonser, L.R.; Eckalbar, W.L.; Rodriguez, L.; Shen, J.; Koh, K.D.; Ghias, K.; Zlock, L.T.; Christenson, S.; Woodruff, P.G.; Finkbeiner, W.E.; et al. The Type 2 Asthma Mediator IL-13 Inhibits SARS-CoV-2 Infection of Bronchial Epithelium. Am. J. Respir. Cell Mol. Biol. 2022, 66, 391–401. [Google Scholar] [CrossRef]
- Hospitalized Adults: Therapeutic Management. Available online: https://www.covid19treatmentguidelines.nih.gov/management/clinical-management-of-adults/hospitalized-adults--therapeutic-management/ (accessed on 7 December 2022).
- RECOVERY Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Asif, S.; Frithiof, R.; Larsson, A.; Franzén, S.; Anderberg, S.B.; Kristensen, B.; Hultström, M.; Lipcsey, M. Immuno-Modulatory Effects of Dexamethasone in Severe COVID-19—A Swedish Cohort Study. Biomedicines 2023, 11, 164. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective Treatment of Severe COVID-19 Patients with Tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, C.; Dolci, G.; Massari, M.; Merlo, D.F.; Cavuto, S.; Savoldi, L.; Bruzzi, P.; Boni, F.; Braglia, L.; Turrà, C.; et al. Effect of Tocilizumab vs Standard Care on Clinical Worsening in Patients Hospitalized with COVID-19 Pneumonia: A Randomized Clinical Trial. JAMA Intern. Med. 2021, 181, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Hermine, O.; Mariette, X.; Tharaux, P.-L.; Resche-Rigon, M.; Porcher, R.; Ravaud, P.; CORIMUNO-19 Collaborative Group. Effect of Tocilizumab vs Usual Care in Adults Hospitalized with COVID-19 and Moderate or Severe Pneumonia: A Randomized Clinical Trial. JAMA Intern. Med. 2021, 181, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Frigault, M.J.; Serling-Boyd, N.J.; Fernandes, A.D.; Harvey, L.; Foulkes, A.S.; Horick, N.K.; Healy, B.C.; Shah, R.; Bensaci, A.M.; et al. Efficacy of Tocilizumab in Patients Hospitalized with COVID-19. N. Engl. J. Med. 2020, 383, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- RECOVERY Collaborative Group. Tocilizumab in Patients Admitted to Hospital with COVID-19 (RECOVERY): A Randomised, Controlled, Open-Label, Platform Trial. Lancet 2021, 397, 1637–1645. [Google Scholar] [CrossRef]
- Cheema, A.H.; Chaludiya, K.; Khalid, M.; Nwosu, M.; Konka, S.; Agyeman, W.Y.; Bisht, A.; Gopinath, A.; Arcia Franchini, A.P. Efficacy of Anakinra in Pericarditis: A Systematic Review. Cureus 2022, 14, e29862. [Google Scholar] [CrossRef] [PubMed]
- Kineret® (Anakinra) for Injection, for Subcutaneous Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/103950s5189lbl.pdf (accessed on 13 June 2023).
- Barkas, F.; Filippas-Ntekouan, S.; Kosmidou, M.; Liberopoulos, E.; Liontos, A.; Milionis, H. Anakinra in Hospitalized Non-Intubated Patients with Coronavirus Disease 2019: A Systematic Review and Meta-Analysis. Rheumatology 2021, 60, 5527–5537. [Google Scholar] [CrossRef]
- Cecchi, L.; Vaghi, A.; Bini, F.; Martini, M.; Musarra, A.; Bilò, M.B. From Triggers to Asthma: A Narrative Review on Epithelium Dysfunction. Eur. Ann. Allergy Clin. Immunol. 2022, 54, 245. [Google Scholar] [CrossRef]
- Schleimer, R.P. An Overview of Glucocorticoid Anti-Inflammatory Actions. Eur. J. Clin. Pharmacol. 1993, 45, S3–S7. [Google Scholar] [CrossRef]
- Barnes, P.J. Inhaled Corticosteroids. Pharmaceuticals 2010, 3, 514–540. [Google Scholar] [CrossRef]
- Schwiebert, L.A.; Beck, L.A.; Stellato, C.; Bickel, C.A.; Bochner, B.S.; Schleimer, R.P. Glucocorticosteroid Inhibition of Cytokine Production: Relevance to Antiallergic Actions. J. Allergy Clin. Immunol. 1996, 97, 143–152. [Google Scholar] [CrossRef]
- Ek, A.; Larsson, K.; Siljerud, S.; Palmberg, L. Fluticasone and Budesonide Inhibit Cytokine Release in Human Lung Epithelial Cells and Alveolar Macrophages. Allergy 1999, 54, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Fukuda, Y.; Uchida, Y.; Uno, T.; Jinno, M.; Kishino, Y.; Yamamoto, M.; Sato, H.; Akimoto, K.; Kaneko, K.; et al. Inhibition of Virus-Induced Cytokine Production from Airway Epithelial Cells by the Late Addition of Budesonide. Medicina 2020, 56, 98. [Google Scholar] [CrossRef] [PubMed]
- Skevaki, C.L.; Christodoulou, I.; Spyridaki, I.S.; Tiniakou, I.; Georgiou, V.; Xepapadaki, P.; Kafetzis, D.A.; Papadopoulos, N.G. Budesonide and Formoterol Inhibit Inflammatory Mediator Production by Bronchial Epithelial Cells Infected with Rhinovirus. Clin. Exp. Allergy 2009, 39, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Rimmer, C.; Hetelekides, S.; Eliseeva, S.I.; Georas, S.N.; Veazey, J.M. Budesonide Promotes Airway Epithelial Barrier Integrity Following Double-Stranded RNA Challenge. PLoS ONE 2021, 16, e0260706. [Google Scholar] [CrossRef]
- Finney, L.J.; Glanville, N.; Farne, H.; Aniscenko, J.; Fenwick, P.; Kemp, S.V.; Trujillo-Torralbo, M.-B.; Loo, S.L.; Calderazzo, M.A.; Wedzicha, J.A.; et al. Inhaled Corticosteroids Downregulate the SARS-CoV-2 Receptor ACE2 in COPD through Suppression of Type I Interferon. J. Allergy Clin. Immunol. 2021, 147, 510–519.e5. [Google Scholar] [CrossRef]
- Yamaya, M.; Nishimura, H.; Deng, X.; Sugawara, M.; Watanabe, O.; Nomura, K.; Shimotai, Y.; Momma, H.; Ichinose, M.; Kawase, T. Inhibitory Effects of Glycopyrronium, Formoterol, and Budesonide on Coronavirus HCoV-229E Replication and Cytokine Production by Primary Cultures of Human Nasal and Tracheal Epithelial Cells. Respir. Investig. 2020, 58, 155–168. [Google Scholar] [CrossRef]
- Matsuyama, S.; Kawase, M.; Nao, N.; Shirato, K.; Ujike, M.; Kamitani, W.; Shimojima, M.; Fukushi, S. The Inhaled Steroid Ciclesonide Blocks SARS-CoV-2 RNA Replication by Targeting the Viral Replication-Transcription Complex in Cultured Cells. J. Virol. 2020, 95, e01648-20. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Nicolau, D.V.; Langford, B.; Mahdi, M.; Jeffers, H.; Mwasuku, C.; Krassowska, K.; Fox, R.; Binnian, I.; Glover, V.; et al. Inhaled Budesonide in the Treatment of Early COVID-19 (STOIC): A Phase 2, Open-Label, Randomised Controlled Trial. Lancet Respir. Med. 2021, 9, 763–772. [Google Scholar] [CrossRef]
- Yu, L.-M.; Bafadhel, M.; Dorward, J.; Hayward, G.; Saville, B.R.; Gbinigie, O.; Van Hecke, O.; Ogburn, E.; Evans, P.H.; Thomas, N.P.B.; et al. Inhaled Budesonide for COVID-19 in People at High Risk of Complications in the Community in the UK (PRINCIPLE): A Randomised, Controlled, Open-Label, Adaptive Platform Trial. Lancet 2021, 398, 843–855. [Google Scholar] [CrossRef]
- Reis, G.; dos Santos Moreira Silva, E.A.; Silva, D.C.M.; Thabane, L.; de Souza Campos, V.H.; Ferreira, T.S.; Quirino dos Santos, C.V.; Ribeiro Nogueira, A.M.; Figueiredo Guimaraes Almeida, A.P.; Cançado Monteiro Savassi, L.; et al. Oral Fluvoxamine with Inhaled Budesonide for Treatment of Early-Onset COVID-19. Ann. Intern. Med. 2023, 176, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, M.E.; Ruddy, M.K.; Pavord, I.D.; Israel, E.; Rabe, K.F.; Ford, L.B.; Maspero, J.F.; Abdulai, R.M.; Hu, C.-C.; Martincova, R.; et al. Efficacy and Safety of Itepekimab in Patients with Moderate-to-Severe Asthma. N. Engl. J. Med. 2021, 385, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Menzies-Gow, A.; Corren, J.; Bourdin, A.; Chupp, G.; Israel, E.; Wechsler, M.E.; Brightling, C.E.; Griffiths, J.M.; Hellqvist, Å.; Bowen, K.; et al. Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma. N. Engl. J. Med. 2021, 384, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.L.; Korenblat, P.E. Anti-Interleukin 4 and 13 for Asthma Treatment in the Era of Endotypes. Curr. Opin. Allergy Clin. Immunol. 2014, 14, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, C.; Pelaia, G.; Longhini, F.; Crimi, C.; Calabrese, C.; Gallelli, L.; Sciacqua, A.; Vatrella, A. Monoclonal Antibodies Targeting Alarmins: A New Perspective for Biological Therapies of Severe Asthma. Biomedicines 2021, 9, 1108. [Google Scholar] [CrossRef] [PubMed]
- Desvaux, E.; Hamon, A.; Hubert, S.; Boudjeniba, C.; Chassagnol, B.; Swindle, J.; Aussy, A.; Laigle, L.; Laplume, J.; Soret, P.; et al. Network-Based Repurposing Identifies Anti-Alarmins as Drug Candidates to Control Severe Lung Inflammation in COVID-19. PLoS ONE 2021, 16, e0254374. [Google Scholar] [CrossRef]
- Ryu, G.; Shin, H.-W. SARS-CoV-2 Infection of Airway Epithelial Cells. Immune Netw. 2021, 21, e3. [Google Scholar] [CrossRef]
- Bridges, J.P.; Vladar, E.K.; Huang, H.; Mason, R.J. Respiratory Epithelial Cell Responses to SARS-CoV-2 in COVID-19. Thorax 2022, 77, 203–209. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, T.J.F.; Singhera, G.K.; Leung, J.M.; Dorscheid, D.R. Airway Epithelial-Derived Immune Mediators in COVID-19. Viruses 2023, 15, 1655. https://doi.org/10.3390/v15081655
Guo TJF, Singhera GK, Leung JM, Dorscheid DR. Airway Epithelial-Derived Immune Mediators in COVID-19. Viruses. 2023; 15(8):1655. https://doi.org/10.3390/v15081655
Chicago/Turabian StyleGuo, Tony J. F., Gurpreet K. Singhera, Janice M. Leung, and Delbert R. Dorscheid. 2023. "Airway Epithelial-Derived Immune Mediators in COVID-19" Viruses 15, no. 8: 1655. https://doi.org/10.3390/v15081655
APA StyleGuo, T. J. F., Singhera, G. K., Leung, J. M., & Dorscheid, D. R. (2023). Airway Epithelial-Derived Immune Mediators in COVID-19. Viruses, 15(8), 1655. https://doi.org/10.3390/v15081655