
Human Post-Translational SUMOylation Modification of SARS-CoV-2 Nucleocapsid Protein Enhances Its Interaction Affinity with Itself and Plays a Critical Role in Its Nuclear Translocation

 and
and
Abstract
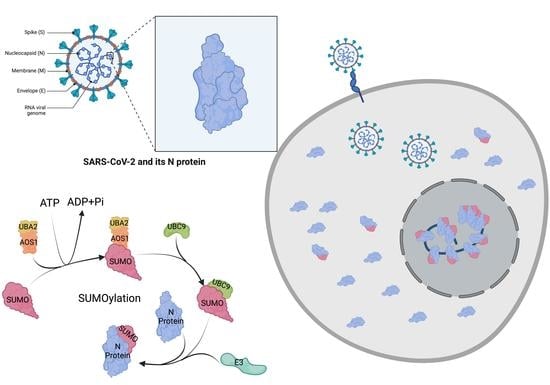
1. Introduction
2. Materials and Methods
2.1. Expression and Purification of SUMOylation Enzymes and SARS-CoV-2 N Protein
2.2. In Vitro SUMOylation Assay
2.3. Mass Spectrometry Analysis to Determine SUMO-Modified Lysine on N Protein
2.4. LTQ Orbitrap Xl Loading and Run
2.5. Bioinformatic Analysis of MS Data
2.6. Validation of SUMOylation Sites of SARS-CoV-2 N Protein Using Engineered SUMO1 Peptide
2.7. Construction and Design of N Protein Lysine to Arginine Mutants
2.8. In Vitro SUMOylation with qFRET Reporter for N Protein Mutants
2.9. KD Determination of SUMOylated or Not-SUMOylated N Proteins Using qFRET
2.10. SARS-CoV-2 N Protein Aggregation Assay with or without SUMO Modification
2.11. Cellular Translocation of N Protein
2.12. Statistical Analysis
3. Results
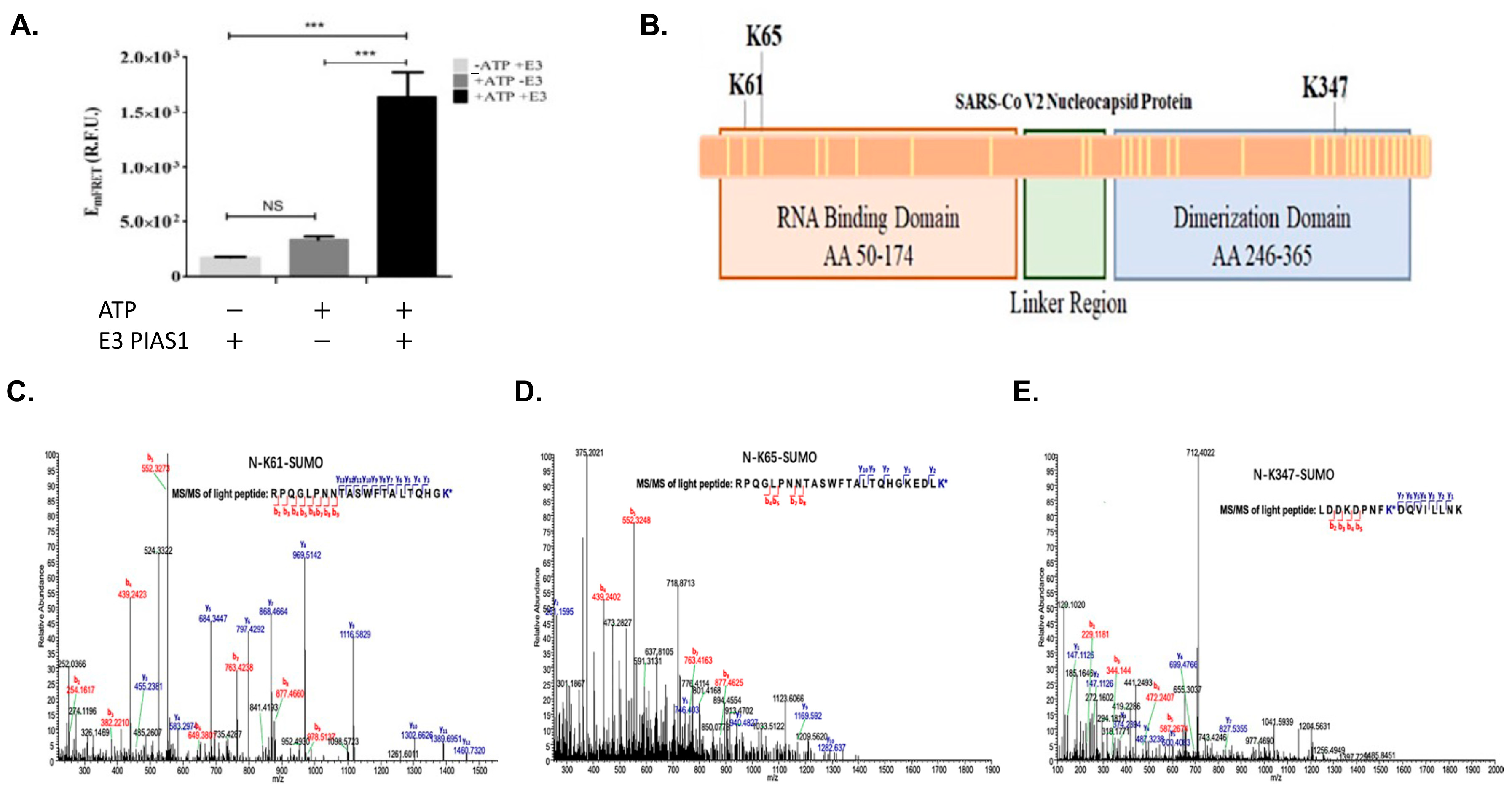
3.1. In Vitro qFRET Assay for SUMOylation of SARS-CoV-2 N Protein
3.2. Mass Spectrometry Analysis to Determine SUMO Modified Lysine on SARS-CoV-2 N Protein
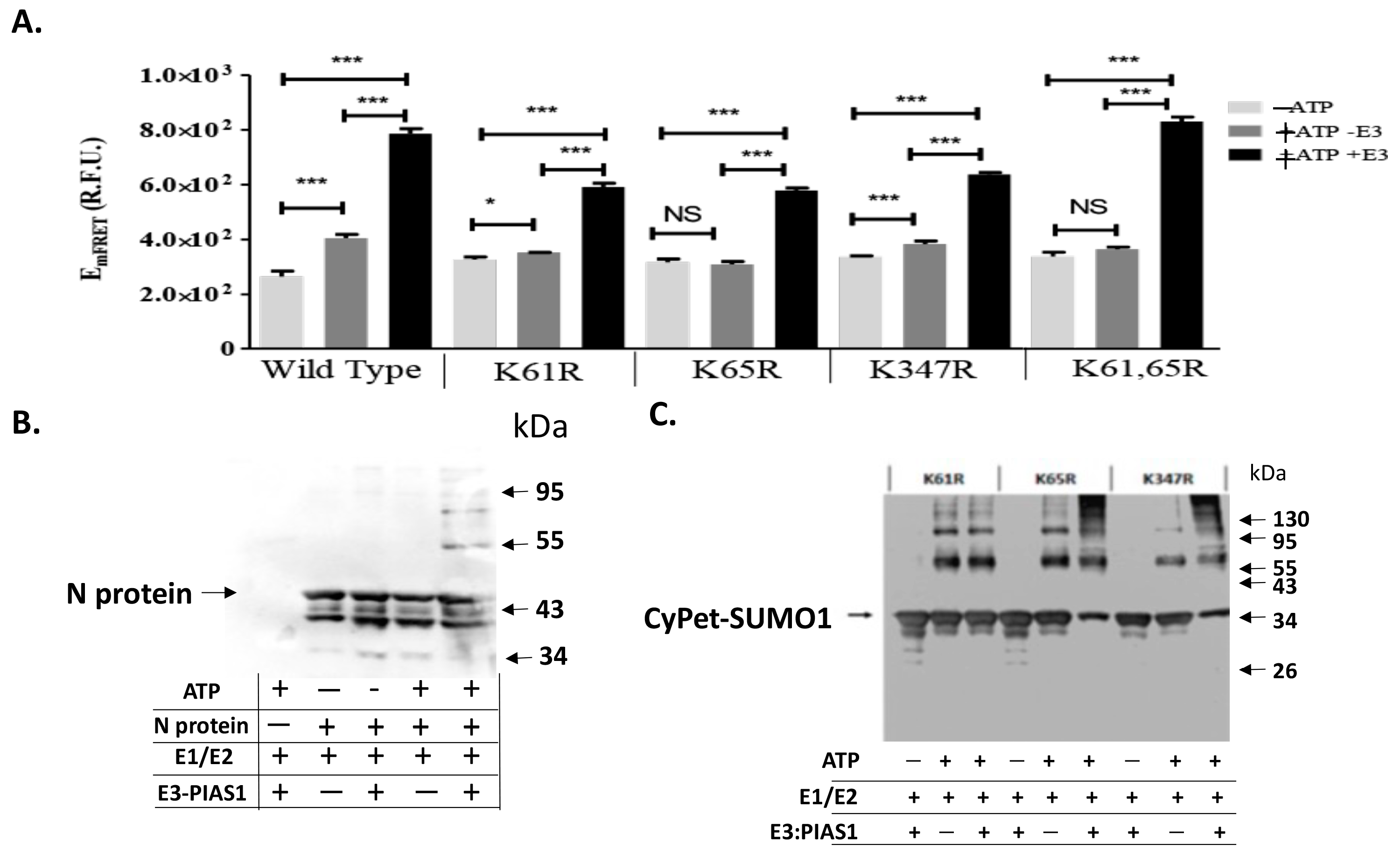
3.3. qFRET Assay for In Vitro SUMOylation Assay of N Protein Mutants
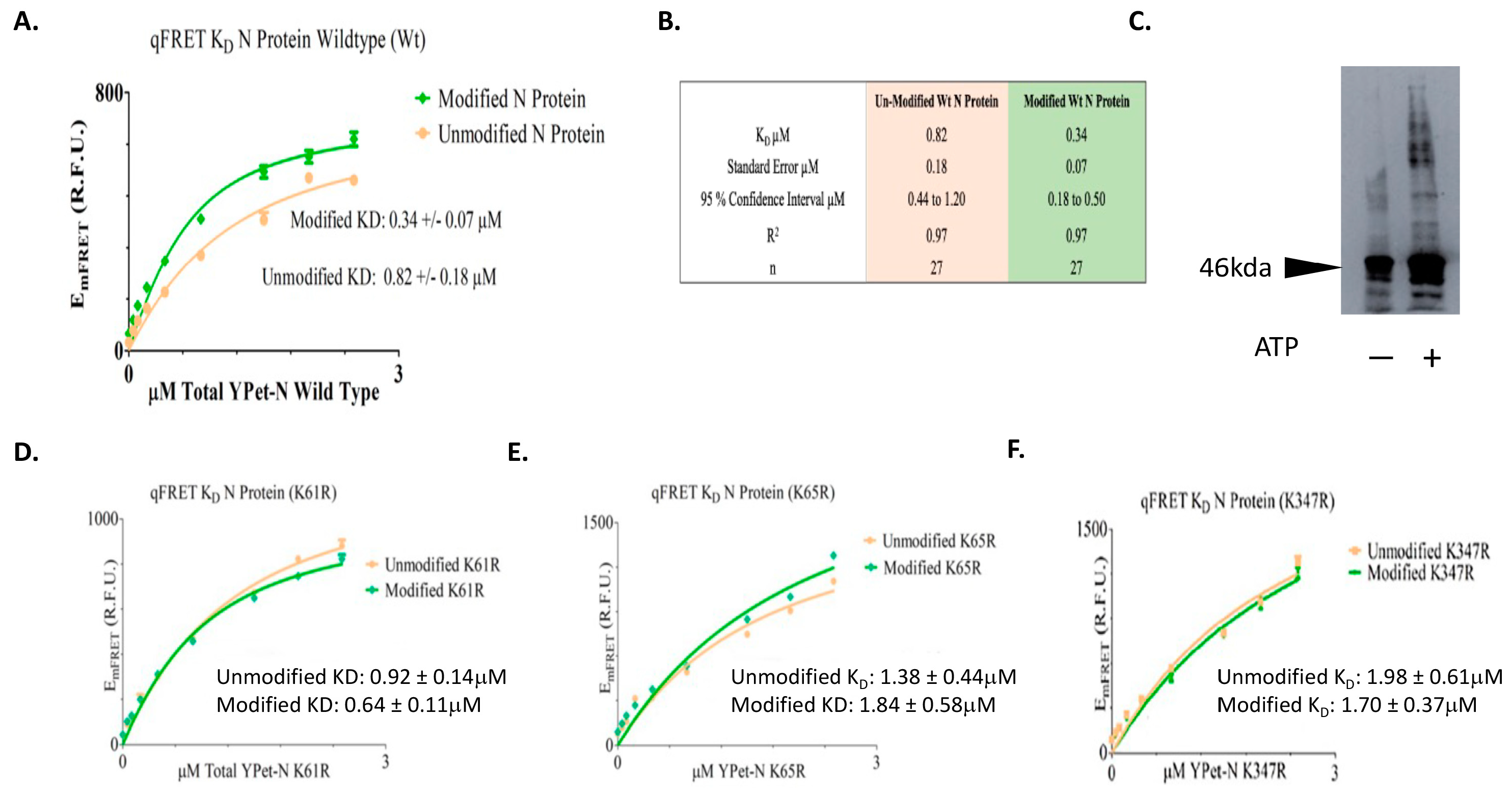
3.4. KD Determination of SUMOylated or Not SUMOylated N Protein Using qFRET
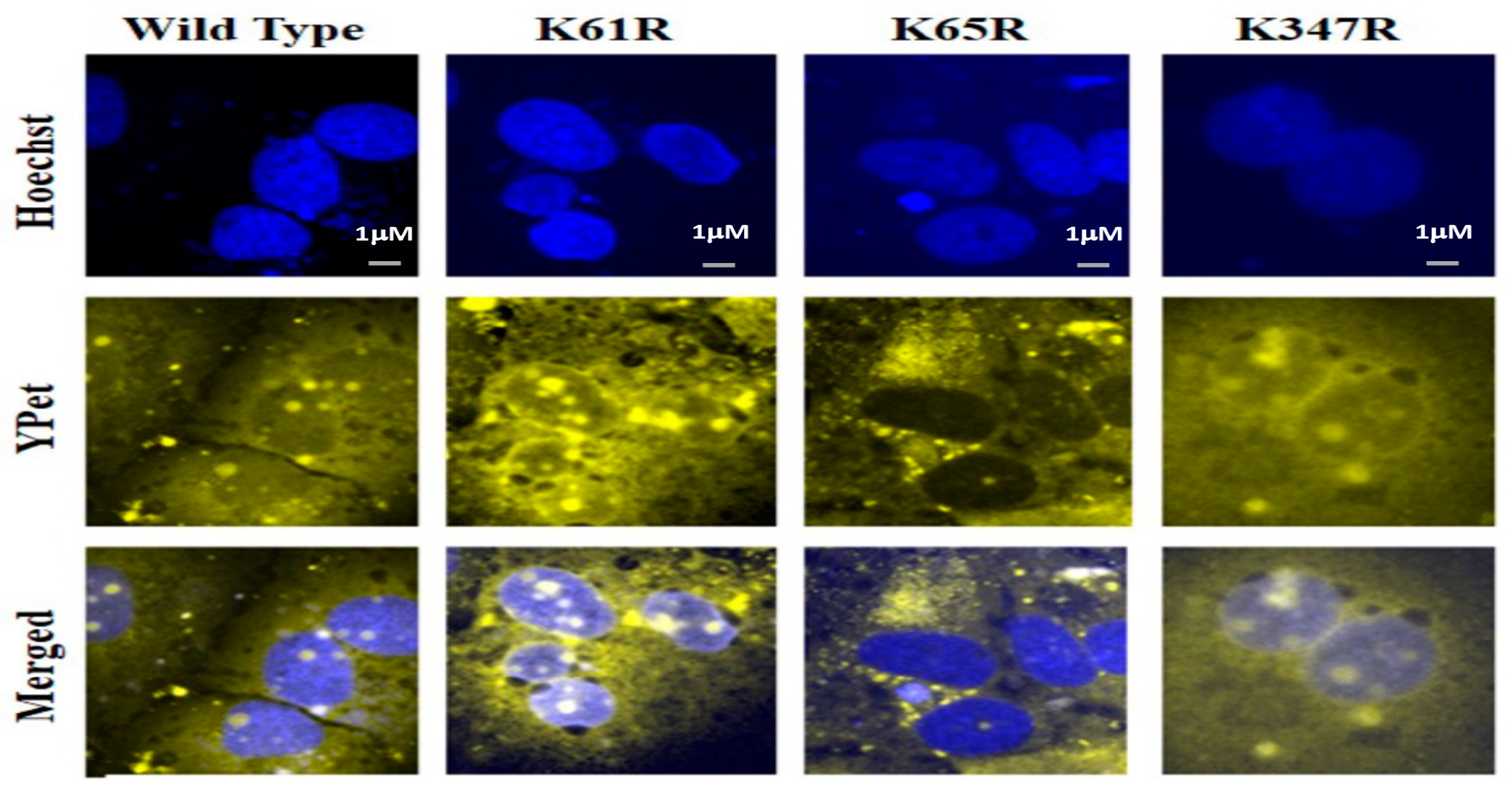
3.5. Nucleus Translocation of N Protein
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- He, R.; Leeson, A.; Ballantine, M.; Andonov, A.; Baker, L.; Dobie, F.; Li, Y.; Bastien, N.; Feldmann, H.; Strocher, U.; et al. Characterization of protein-protein interactions between the nucleocapsid protein and membrane protein of the SARS coronavirus. Virus Res. 2004, 105, 121–125. [Google Scholar] [CrossRef]
- Luo, H.; Chen, J.; Chen, K.; Shen, X.; Jiang, H. Carboxyl terminus of severe acute respiratory syndrome coronavirus nucleocapsid protein: Self-association analysis and nucleic acid binding characterization. Biochemistry 2006, 45, 11827–11835. [Google Scholar] [CrossRef]
- Chang, C.K.; Hou, M.H.; Chang, C.F.; Hsiao, C.D.; Huang, T.H. The SARS coronavirus nucleocapsid protein--forms and functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R., III; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef]
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Safari, M.; Frankel, A.D.; Morgan, D.O. Phosphoregulation of Phase Separation by the SARS-CoV-2 N Protein Suggests a Biophysical Basis for its Dual Functions. Mol. Cell 2020, 80, 1092–1103.e1094. [Google Scholar] [CrossRef]
- Savastano, A.; Ibanez de Opakua, A.; Rankovic, M.; Zweckstetter, M. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat. Commun. 2020, 11, 6041. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 183, 730–738.e713. [Google Scholar] [CrossRef]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortola, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef] [PubMed]
- Tung, H.Y.L.; Limtung, P. Mutations in the phosphorylation sites of SARS-CoV-2 encoded nucleocapsid protein and structure model of sequestration by protein 14-3-3. Biochem. Biophys. Res. Commun. 2020, 532, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Kumar, R.; Mishra, R.N.; Reddy, M.K.; Chow, V.T.; Lal, S.K. The severe acute respiratory syndrome coronavirus nucleocapsid protein is phosphorylated and localizes in the cytoplasm by 14-3-3-mediated translocation. J. Virol. 2005, 79, 11476–11486. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Dove, B.K.; Enjuanes, L.; DeDiego, M.L.; Alvarez, E.; Howell, G.; Heinen, P.; Zambon, M.; Hiscox, J.A. Subcellular localization of the severe acute respiratory syndrome coronavirus nucleocapsid protein. J. Gen. Virol. 2005, 86 Pt 12, 3303–3310. [Google Scholar] [CrossRef]
- Timani, K.A.; Liao, Q.; Ye, L.; Zeng, Y.; Liu, J.; Zheng, Y.; Ye, L.; Yang, X.; Lingbao, K.; Gao, J.; et al. Nuclear/nucleolar localization properties of C-terminal nucleocapsid protein of SARS coronavirus. Virus Res. 2005, 114, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Lal, S.K. The SARS-CoV nucleocapsid protein: A protein with multifarious activities. Infect. Genet. Evol. 2008, 8, 397–405. [Google Scholar] [CrossRef]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Fang, Y.; Yang, Q.; Shu, T.; Wang, A.; Huang, M.; Jin, L.; Deng, F.; Qiu, Y.; Zhou, X. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 2020, 6, 65. [Google Scholar] [CrossRef]
- Li, F.Q.; Xiao, H.; Tam, J.P.; Liu, D.X. Sumoylation of the nucleocapsid protein of severe acute respiratory syndrome coronavirus. FEBS Lett. 2005, 579, 2387–2396. [Google Scholar] [CrossRef]
- Sun, Z.; Ren, K.; Zhang, X.; Chen, J.; Jiang, Z.; Jiang, J.; Ji, F.; Ouyang, X.; Li, L. Mass Spectrometry Analysis of Newly Emerging Coronavirus HCoV-19 Spike Protein and Human ACE2 Reveals Camouflaging Glycans and Unique Post-Translational Modifications. Engineering 2021, 7, 1441–1451. [Google Scholar] [CrossRef]
- Miyawaki, A. Development of probes for cellular functions using fluorescent proteins and fluorescence resonance energy transfer. Annu. Rev. Biochem. 2011, 80, 357–373. [Google Scholar] [CrossRef]
- Sun, Y.; Wallrabe, H.; Seo, S.A.; Periasamy, A. FRET microscopy in 2010: The legacy of Theodor Forster on the 100th anniversary of his birth. Chemphyschem 2011, 12, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.G.; Alseikhan, B.A.; Peterson, B.Z.; Yue, D.T. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron 2001, 31, 973–985. [Google Scholar] [CrossRef]
- Martin, S.F.; Tatham, M.H.; Hay, R.T.; Samuel, I.D. Quantitative analysis of multi-protein interactions using FRET: Application to the SUMO pathway. Protein Sci. 2008, 17, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Hoppe, A.D.; Kainkaryam, R.; Woolf, P.J.; Linderman, J.J. A computational approach to inferring cellular protein-binding affinities from quantitative fluorescence resonance energy transfer imaging. Proteomics 2009, 9, 5371–5383. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Madahar, V.; Liao, J. Development of FRET Assay into Quantitative and High-throughput Screening Technology Platforms for Protein-Protein Interactions. Ann. Biomed. Eng. 2011, 39, 1224–1234. [Google Scholar] [CrossRef]
- Song, Y.; Rodgers, V.G.; Schultz, J.S.; Liao, J. Protein interaction affinity determination by quantitative FRET technology. Biotechnol. Bioeng. 2012, 109, 2875–2883. [Google Scholar] [CrossRef]
- Jiang, L.; Xiong, Z.; Song, Y.; Lu, Y.; Chen, Y.; Schultz, J.S.; Li, J.; Liao, J. Protein-Protein Affinity Determination by Quantitative FRET Quenching. Sci. Rep. 2019, 9, 2050. [Google Scholar] [CrossRef]
- Liao, J.; Madahar, V.; Dang, R.; Jiang, L. Quantitative FRET (qFRET) Technology for the Determination of Protein-Protein Interaction Affinity in Solution. Molecules 2021, 26, 6339. [Google Scholar] [CrossRef]
- Merrill, J.C.; Melhuish, T.A.; Kagey, M.H.; Yang, S.H.; Sharrocks, A.D.; Wotton, D. A role for non-covalent SUMO interaction motifs in Pc2/CBX4 E3 activity. PLoS ONE 2010, 5, e8794. [Google Scholar] [CrossRef]
- Arriagada, G.; Muntean, L.N.; Goff, S.P. SUMO-interacting motifs of human TRIM5alpha are important for antiviral activity. PLoS Pathog. 2011, 7, e1002019. [Google Scholar] [CrossRef]
- Ptak, C.; Wozniak, R.W. SUMO and Nucleocytoplasmic Transport. Adv. Exp. Med. Biol. 2017, 963, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A. Interplay between nuclear transport and ubiquitin/SUMO modifications in the regulation of cancer-related proteins. Semin Cancer Biol. 2014, 27, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Xie, Y.; Zheng, Y.; Jiang, S.; Liu, W.; Mu, W.; Liu, Z.; Zhao, Y.; Xue, Y.; Ren, J. GPS-SUMO: A tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res. 2014, 42, W325–W330. [Google Scholar] [CrossRef]
- Beauclair, G.; Bridier-Nahmias, A.; Zagury, J.F.; Saib, A.; Zamborlini, A. JASSA: A comprehensive tool for prediction of SUMOylation sites and SIMs. Bioinformatics 2015, 31, 3483–3491. [Google Scholar] [CrossRef]
- Prussia, A.; Thepchatri, P.; Snyder, J.P.; Plemper, R.K. Systematic approaches towards the development of host-directed antiviral therapeutics. Int. J. Mol. Sci. 2011, 12, 4027–4052. [Google Scholar] [CrossRef]
- de Chassey, B.; Meyniel-Schicklin, L.; Aublin-Gex, A.; Andre, P.; Lotteau, V. New horizons for antiviral drug discovery from virus-host protein interaction networks. Curr. Opin. Virol. 2012, 2, 606–613. [Google Scholar] [CrossRef]
- Ma-Lauer, Y.; Lei, J.; Hilgenfeld, R.; von Brunn, A. Virus-host interactomes--antiviral drug discovery. Curr. Opin. Virol. 2012, 2, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.; Ranjan, K.; Brar, B.; Shah, I.; Lalmbe, U.; Manimegalai, J.; Vashisht, B.; Gaury, M.; Kumar, P.; Khurana, S.K.; et al. Virus-Host Interactions: New Insights and Advances in Drug Development Against Viral Pathogens. Curr. Drug Metab. 2017, 18, 942–970. [Google Scholar] [CrossRef]
- Liao, J.; Way, G.; Madahar, V. Target Virus or Target Ourselves for COVID-19 Drugs Discovery?-Lessons learned from anti-influenzas virus therapies. Med. Drug Discov. 2020, 5, 100037. [Google Scholar] [CrossRef]
- Wimmer, P.; Schreiner, S.; Dobner, T. Human pathogens and the host cell SUMOylation system. J. Virol. 2012, 86, 642–654. [Google Scholar] [CrossRef]
- Everett, R.D.; Boutell, C.; Hale, B.G. Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 2013, 11, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Lowrey, A.J.; Cramblet, W.; Bentz, G. Viral manipulation of the cellular sumoylation machinery. Cell Commun. Signal. 2017, 15, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Kim, K.H.; Makino, S. Characterization of N protein self-association in coronavirus ribonucleoprotein complexes. Virus Res. 2003, 98, 131–140. [Google Scholar] [CrossRef]
- Surjit, M.; Liu, B.; Kumar, P.; Chow, V.T.; Lal, S.K. The nucleocapsid protein of the SARS coronavirus is capable of self-association through a C-terminal 209 amino acid interaction domain. Biochem. Biophys. Res. Commun. 2004, 317, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.M.; Gustafson, C.L.; Diao, J.; Burgner, J.W., II; Li, Z.; Zhang, J.; Chen, J. Recombinant severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein forms a dimer through its C-terminal domain. J. Biol. Chem. 2005, 280, 23280–23286. [Google Scholar] [CrossRef]
- Rowland, R.R.; Chauhan, V.; Fang, Y.; Pekosz, A.; Kerrigan, M.; Burton, M.D. Intracellular localization of the severe acute respiratory syndrome coronavirus nucleocapsid protein: Absence of nucleolar accumulation during infection and after expression as a recombinant protein in vero cells. J. Virol. 2005, 79, 11507–11512. [Google Scholar] [CrossRef]
- Matunis, M.J.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 1996, 135 Pt 1, 1457–1470. [Google Scholar] [CrossRef]
- Erazo, T.; Espinosa-Gil, S.; Dieguez-Martinez, N.; Gomez, N.; Lizcano, J.M. SUMOylation Is Required for ERK5 Nuclear Translocation and ERK5-Mediated Cancer Cell Proliferation. Int. J. Mol. Sci. 2020, 21, 2203. [Google Scholar] [CrossRef]
- Terui, Y.; Saad, N.; Jia, S.; McKeon, F.; Yuan, J. Dual role of sumoylation in the nuclear localization and transcriptional activation of NFAT1. J. Biol. Chem. 2004, 279, 28257–28265. [Google Scholar] [CrossRef]
- Du, J.X.; Bialkowska, A.B.; McConnell, B.B.; Yang, V.W. SUMOylation regulates nuclear localization of Kruppel-like factor 5. J. Biol. Chem. 2008, 283, 31991–32002. [Google Scholar] [CrossRef]
- Liao, J.Y.; Song, Y.; Liu, Y. A new trend to determine biochemical parameters by quantitative FRET assays. Acta Pharmacol. Sin. 2015, 36, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Way, G.; Xiong, Z.; Wang, G.; Dai, H.; Zheng, S.; Garcia-Sastre, A.; Liao, J. A novel SUMOylation site in the influenza a virus NS1 protein identified with a highly sensitive FRET assay. J. Biotechnol. 2020, 323, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Kaci, A.; Keindl, M.; Solheim, M.H.; Njolstad, P.R.; Bjorkhaug, L.; Aukrust, I. The E3 SUMO ligase PIASgamma is a novel interaction partner regulating the activity of diabetes associated hepatocyte nuclear factor-1alpha. Sci. Rep. 2018, 8, 12780. [Google Scholar] [CrossRef] [PubMed]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Polo, S.; Miller, K.M.; Jackson, S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 2009, 462, 935–939. [Google Scholar] [CrossRef]
- Chanda, A.; Chan, A.; Deng, L.; Kornaga, E.N.; Enwere, E.K.; Morris, D.G.; Bonni, S. Identification of the SUMO E3 ligase PIAS1 as a potential survival biomarker in breast cancer. PLoS ONE 2017, 12, e0177639. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.W.; Daugherty, P.S. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 2005, 23, 355–360. [Google Scholar] [CrossRef]
- Algar, W.R.; Hildebrandt, N.; Vogel, S.S.; Medintz, I.L. FRET as a biomolecular research tool-understanding its potential while avoiding pitfalls. Nat. Methods 2019, 16, 815–829. [Google Scholar] [CrossRef]
- Chen, T.; He, B.; Tao, J.; He, Y.; Deng, H.; Wang, X.; Zheng, Y. Application of Forster Resonance Energy Transfer (FRET) technique to elucidate intracellular and In Vivo biofate of nanomedicines. Adv. Drug Deliv. Rev. 2019, 143, 177–205. [Google Scholar] [CrossRef]
- Liu, Y.; Song, Y.; Jiang, L.; Liao, J. Quantitative analysis of FRET assay in biology. Front. Biol. 2012, 7, 57–64. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N Protein | K61R Unmodified | K61R Modified | K65R Unmodified | K65R Modified | K347R Unmodified | K347R Modified |
|---|---|---|---|---|---|---|
| KD (µM) | 0.92 | 0.64 | 1.38 | 1.84 | 1.98 | 1.70 |
| Standard Error (µM) | 0.14 | 0.11 | 0.44 | 0.58 | 0.61 | 0.37 |
| 95 % Confidence Interval (µM) | 0.62 to 1.21 | 0.41 to 0.87 | 0.46 to 2.30 | 0.64 to 3.03 | 0.71 to 3.21 | 0.99 to 3.97 |
| R2 | 0.99 | 0.98 | 0.95 | 0.91 | 0.97 | 0.97 |
| n | 27 | 27 | 27 | 27 | 27 | 27 |
| pET28B Primers | |
|---|---|
| K61Rfor | ccagcatggcagagaagacctgaaattt |
| K61Rrev | caggtcttctctgccatgctgggtcag |
| K65Rfor | gaagacctgagatttccgcgcggccag |
| K65Rrev | ctggccgcgcggaaatctcaggtcttc |
| K347Rfor | gatccgaattttcgagatcaggtgatt |
| K347Rrev | aatcacctgatctcgaaaattcggatc |
| pCDNA3.1 Primers | |
|---|---|
| pcD_Ncwt_61For | ctcactcaacatggcagggaagacctt |
| pcD_Ncwt_61Rev | aaggtcttccctgccatgttgagtgag |
| pcD_Ncwt_65For | gaagaccttagattccctcgaggacaa |
| pcD_Ncwt_65Rev | ttgtcctcgagggaatctaaggtcttc |
| pcD_Ncwt_347For | aaagatccaaatttcagagatcaagtcatt |
| pcD_Ncwt_347Rev | aatgacttgatctctgaaatttggatcttt |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madahar, V.; Dang, R.; Zhang, Q.; Liu, C.; Rodgers, V.G.J.; Liao, J. Human Post-Translational SUMOylation Modification of SARS-CoV-2 Nucleocapsid Protein Enhances Its Interaction Affinity with Itself and Plays a Critical Role in Its Nuclear Translocation. Viruses 2023, 15, 1600. https://doi.org/10.3390/v15071600
Madahar V, Dang R, Zhang Q, Liu C, Rodgers VGJ, Liao J. Human Post-Translational SUMOylation Modification of SARS-CoV-2 Nucleocapsid Protein Enhances Its Interaction Affinity with Itself and Plays a Critical Role in Its Nuclear Translocation. Viruses. 2023; 15(7):1600. https://doi.org/10.3390/v15071600
Chicago/Turabian StyleMadahar, Vipul, Runrui Dang, Quanqing Zhang, Chuchu Liu, Victor G. J. Rodgers, and Jiayu Liao. 2023. "Human Post-Translational SUMOylation Modification of SARS-CoV-2 Nucleocapsid Protein Enhances Its Interaction Affinity with Itself and Plays a Critical Role in Its Nuclear Translocation" Viruses 15, no. 7: 1600. https://doi.org/10.3390/v15071600
APA StyleMadahar, V., Dang, R., Zhang, Q., Liu, C., Rodgers, V. G. J., & Liao, J. (2023). Human Post-Translational SUMOylation Modification of SARS-CoV-2 Nucleocapsid Protein Enhances Its Interaction Affinity with Itself and Plays a Critical Role in Its Nuclear Translocation. Viruses, 15(7), 1600. https://doi.org/10.3390/v15071600