Abstract
Transmissible spongiform encephalopathies (TSEs) or prion diseases are characterized by the accumulation in affected tissues of the abnormal prion protein PrPTSE. We previously demonstrated PrPTSE in the blood of macaques experimentally infected with variant Creutzfeldt–Jakob disease (vCJD), a human TSE, months to years prior to clinical onset. That work supported the prospect of using PrPTSE as a blood biomarker to detect vCJD and possibly other human TSEs before the onset of overt illness. However, our results also raised questions about the origin of PrPTSE detected in blood early after inoculation and the effects of dose and route on the timing of the appearance of PrPTSE. To investigate these questions, we inoculated vCJD-susceptible transgenic mice and non-infectable prion protein-knockout mice under inoculation conditions resembling those used in macaques, with additional controls. We assayed PrPTSE in mouse blood using the protein misfolding cyclic amplification (PMCA) method. PrPTSE from the inoculum cleared from the blood of all mice before 2 months post-inoculation (mpi). Mouse PrPTSE generated de novo appeared in blood after 2 mpi. These results were consistent regardless of dose or inoculation route. We also demonstrated that a commercial ELISA-like PrPTSE test detected and quantified PMCA products and provided a useful alternative to Western blots.
1. Introduction
Transmissible spongiform encephalopathies (TSEs or prion diseases) are rare, fatal neurodegenerative disorders of animals and humans. TSEs are characterized by the accumulation in affected tissues of the abnormally folded relatively protease-resistant prion protein named PrPTSE. TSEs have long asymptomatic incubation periods during which infected individuals might donate potentially contaminated biological materials such as blood, tissues, and cells for therapeutic purposes [1]. Variant Creutzfeldt–Jakob disease (vCJD) is a human TSE most often acquired from consumption of food contaminated with the agent that causes bovine spongiform encephalopathy (BSE) in cattle [2,3,4]. Measures to stop the spread of BSE and vCJD were implemented worldwide, and as a result, very few cases of BSE and no new cases of vCJD have been reported in the past few years [5,6]. Among other human TSEs, sporadic CJD (sCJD) is the most common; familial CJD (fCJD) is extremely rare. Family members with certain mutations in the prion protein-encoding (PRNP) gene have an increased risk of developing CJD [7,8].
PrPTSE is currently the only known TSE biomarker; thus, it is the target for all biochemical diagnostic tests under development. The most promising PrPTSE detection methods are protein misfolding cyclic amplification (PMCA) [9] and real-time quaking-induced conversion (RT-QuIC) [10]. Both are in vitro methods that amplify PrPTSE molecules and allow their detection even when PrPTSE is present in extremely low levels such as in biological samples [9,10,11]. PMCA amplified PrPTSE from blood of individuals with vCJD [12,13,14]. In another study, PrPTSE in the blood of vCJD patients was detected using a selective capture method without amplification [15]. Detection of PrPTSE, and infectivity, has yielded inconsistent results in various studies with sCJD blood [13,14,16]. Although new biomarkers and diagnostic tests have been developed to improve CJD diagnostics [7], the field still lacks a reliable, rapid, specific, and sensitive blood-based test to identify humans with TSEs during the preclinical phase of infection. Early detection of PrPTSE would benefit individuals with sCJD and members of families with a history of fCJD by allowing therapeutic trials to be initiated before neurodegeneration becomes severe and irreversible. An assay that identifies persons incubating sCJD might also be useful to screen living donors of human cellular and tissue-derived products, reducing the risk of iatrogenic CJD [1,7].
Several years ago, we began a study to develop a method to detect PrPTSE in the blood of vCJD-infected macaques as a prototype diagnostic test for human vCJD and possibly sCJD [17]. The animals were infected peripherally (intravenous (IV) and intraperitoneal (IP) administration). We generated longitudinal panels of infected animal blood used to track the appearance of PrPTSE over time correlated with the onset of overt (“clinical”) illness [18]. PrPTSE, detected by PMCA, appeared in the blood of cynomolgus macaques as early as 2 months post-inoculation (2 mpi)—the earliest time tested. This unexpected early detection of PrPTSE occurred long before the onset of clinical illness in macaques: 25.5 months for two macaques and 21 months for one macaque before clinical onset [18]. When we conducted a similar study with rhesus macaques injected intraperitoneally with 10 times less vCJD agent compared to the dose used with cynomolgus macaques, PrPTSE appeared in blood 5 and 11 months before clinical onset in two animals, while PrPTSE was not reproducibly detected in one rhesus until 1 month after clinical signs had begun [18]. Although these results confirmed and supported the possible use of PrPTSE as a future preclinical diagnostic biomarker in blood, they also left some unanswered questions that required further investigations. Specifically, we needed to establish whether PrPTSE detected 2 mpi was truly generated de novo as a response to infection or was simply recovered from the residual circulating inoculum. Next, we wanted to assess possible causes of the dramatic difference in the time of appearance of detectable PrPTSE in the blood of rhesus and cynomolgus macaques. To address this latter goal, we focused on the different doses of inoculum injected into the two animal species.
Those basic questions were more feasibly addressed using mice instead of macaques. We used TgBo110 transgenic mice [19] because they are highly susceptible to infection with the vCJD agent and have been used extensively by us and others to study BSE and vCJD [18,19,20,21]. We also used PrP-knockout mice resistant to TSE infections [22] to track clearance of the inoculum without the confounding effects of PrPTSE produced by the infected mice. We attempted to mimic the conditions we previously used to infect macaques with the goal of determining when mouse-generated PrPTSE first appeared in blood and whether the time of appearance was delayed in mice inoculated under conditions matching those used to infect rhesus macaques. Previous studies detected PrPTSE in the blood of rodents experimentally infected with TSEs [23,24], but those studies did not account for PrPTSE remaining from the inoculum. Our studies addressed this consideration. We also explored how the route of inoculation (IV, IP, and intracerebral (IC)—the most commonly studied routes of experimental TSE infection) affected the time when PrPTSE was first detected in blood. Furthermore, we introduced a rapid method to detect PrPTSE products generated by PMCA that could, in some cases, substitute for the Western immunoblots, simplifying test readout and improving throughput. We believe that our data will contribute to the future development and validation of TSE assays based on PrPTSE detection in blood.
2. Materials and Methods
2.1. Mouse Models and Inoculations
TgBo110 mice overexpressing normal bovine prion protein (called “cellular” PrP or PrPC) approximately 8-fold compared to PrPC in brains of cows [19] were originally derived at the Centro de Investigación en Sanidad Animal, Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (CISA-INIA, Madrid, Spain). PrP-KO mice FVB Prnp−/− on FVB/NJ genetic background were kindly donated by Jaroslav Vostal [25] (U.S. Food and Drug Administration, White Oak, Silver Spring) MD, USA. Both mouse strains were bred at the U.S. Food and Drug Administration and confirmed by PCR to have the expected genotypes.
To reduce the volumes of inocula injected into mice to match the inoculation conditions previously used to infect cynomolgus and rhesus macaques [18], we used ratios of circulating blood volumes of macaques and mice. The average volume of circulating blood in adult cynomolgus and rhesus macaques is about 60 mL/kg [26], and the average weight of our macaques at the time of inoculation was 7.5 kg, yielding an estimated average of 450 mL of blood in a macaque. The average volume of circulating blood in a young adult mouse is 72 mL/kg [26], and our mice weighed on average 26 g at inoculation; thus, the average volume of circulating blood in our mice was estimated to be about 1.8 mL. Therefore, we used a 250:1 ratio to calculate appropriate inoculation doses for mice for all inoculation routes. We inoculated groups of 6 to 10 TgBo110 mice and groups of 10 PrP-KO mice by 3 different routes (IV, IP, and IC) and a combination of routes with a homogenate of brain tissue from a cynomolgus macaque with histopathologically confirmed vCJD. We inoculated mice intracerebrally with 30 µL of 1% brain homogenate [18,20]. The 6 inoculation conditions tested and the volumes of inoculum injected are shown in Table 1.

Table 1.
Summary of inoculation conditions for TgBo110 and PrP-knockout mice.
2.2. Blood and Tissue Collections
We collected blood 1 week after inoculation of TgBo110 mice and then every month post-inoculation (mpi) for 2 years until the mice were euthanized. We drew blood from PrP-KO mice using the same schedule but terminated collections at 7 mpi because these mice were not expected to develop TSE. We collected blood by cutting the submandibular vein, obtaining 60–100 µL of citrated blood from each mouse at each time point; we pooled the blood from mice in each inoculation group to obtain sufficient volumes of sample to conduct 2 PMCA tests and immediately stored aliquots of pooled blood at −80 °C. Mice were euthanized when signs such as loss of body weight, scruffy hair coat, and lethargy were observed or when requested by veterinary staff for reasons of animal welfare. We collected approximately 1 mL of citrated blood from each mouse (not pooled) by cardiac puncture before euthanasia and necropsy. The brain and spleen were harvested at necropsy and frozen for further testing. Brain tissue to detect PrPTSE was always excised from the hemisphere opposite the site of injection. Blood, spleens, and brains were assayed for PrPTSE by PMCA.
2.3. Detection of PrPTSE in Mouse Blood by PMCA
As previously described [18], we tested 250 µL aliquots of citrated whole blood treated with a final concentration of 20 U/mL of benzonase nuclease (Millipore-Sigma, Burlington MA, USA) for 30 min at room temperature with constant mixing. Next, we added 250 µL of 20% molecular-grade Sarkosyl (Sigma-Aldrich, St. Louis, MO, USA) and incubated the mixture at room temperature for 30 min with mixing. We ultracentrifuged samples (1 h at 100,000× g, 10 °C), discarded supernatants, and resuspended pellets in 100 µL of a 10% homogenate (PMCA “substrate”) of brains of red-backed voles (bred in-house) [27] in buffer, 50 mL of which contained 0.5 g laboratory-grade Triton X100 (Sigma-Aldrich, St. Louis, MO, USA), 1.5 mL of 5 M molecular-grade NaCl, and 48 mL PBS without calcium and magnesium, pH 7.2; 1 protease inhibitor cocktail tablet containing EDTA (Roche, Sigma-Aldrich, St. Louis, MO, USA) was added immediately prior to use. We transferred the resuspended pellets to 0.2 mL tubes containing three 3/32 in. PTFE beads (McMaster-CARR, Elmhurst, IL, USA) and conducted PMCA using a titanium cuphorn in a programmable Misonix Q700 sonicator (QSonica, Newtown, CT, USA) filled with water and kept inside an incubator set at 37 °C. Each cycle of sonication comprised 10 s pulses every 15 min. The first round of sonication lasted a total of 72 h. After the first round of sonication, we diluted samples 1:10 with fresh vole brain homogenate substrate to conduct round 2 of PMCA and repeated the procedure for round 3; rounds 2 and 3 each lasted 24 h.
To detect PrPTSE in PMCA products by Western blot, we treated aliquots of samples after PMCA, with 50 µg/mL proteinase K for 1 h at 37 °C to remove normal prion protein and denaturing NuPAGE LDS sample buffer (4×) with NuPAGE sample-reducing agent heated at 70 °C for 10 min; we then separated proteins using SDS-PAGE on 12% Bis-Tris precast gels (Invitrogen-Thermo Fisher Scientific, Waltham, MA, USA). We detected prion protein transferred to PVDF membranes using mouse anti-PrP monoclonal antibody 6D11 at a concentration of 6 mg/mL (Research Foundation for Mental Hygiene, New York State Institute for Basic Research) diluted 1:5000 and an HRP-conjugated secondary antibody and then captured chemiluminescent signals on the membranes using the Bio-Rad ChemiDoc Imaging System (Bio-Rad, Hercules, CA, USA).
2.4. Detection of PMCA-Generated Products Using a Rapid PrPTSE Immunoassay
We measured levels of PMCA products using a colorimetric enzyme-linked immunosorbent assay (ELISA-like) called IDEXX HerdChek BSE-Scrapie Antigen Test kit (HC PrPTSE test) that detects PrPTSE as previously described [18]. For this assay, we used 25 µL aliquots of samples collected after rounds 2 and 3 of PMCA, 25 µL of working buffer from the assay kit, and PBS to a total volume of 100 µL. We performed HC assays according to the manufacturer’s instructions (IDEXX Laboratories, Westbrook, ME, USA). We analyzed only rounds 2 and 3 because PrPTSE was undetectable by both Western blot and HC tests after the first round of PMCA.
2.5. Detection of PrPTSE in Mouse Spleen and Brain by PMCA
We gently homogenized 50 mg samples of thawed spleen tissue using sterile disposable pestles in 0.5 mL of PBS. Unhomogenized tissue was discarded, and the suspension was treated with 50 U/mL of benzonase nuclease for 30 min at room temperature with constant agitation. Next, we added 0.5 mL of 20% molecular-grade Sarkosyl to each sample and incubated it at room temperature for 30 min with agitation. We performed ultracentrifugation, PMCA, and Western blot analyses as described above for mouse blood.
We homogenized 10% mouse brain in PBS using a Mini Beadbeater (Biospec Products, Bartlesville, OK, USA) twice for 1.5 min followed by cooling for 5 min on ice. We tested each mouse brain for PrPTSE using PMCA with 25 µL of 10% brain homogenate in 75 µL of normal vole brain homogenate. PMCA and Western blots were performed as described above.
3. Results
3.1. Mouse Inoculations
We inoculated six groups of TgBo110 mice under conditions indicated in Table 1 and further described in Figure S1 in Supplementary Materials. The first two conditions resembled those used to inoculate cynomolgus and rhesus macaques, respectively. Notably, the only difference in inoculations of the two macaque species was that rhesus macaques received 10 times less inoculum by the IP route (IP-low) compared to that injected into cynomolgus macaques by that route (IP-high). IV inoculations were the same for all macaques. We inoculated mice by the IC route—a route not used to inject macaques—to serve as a positive control since we knew from previous studies that IC inoculation of this material at the dilution used should infect all TgBo110 mice. To complete the study, we also injected TgBo110 mice by either the IV or IP route separately. The latter route was also tested at two concentrations of brain suspension, high and low, to match the different concentrations used to inoculate cynomolgus and rhesus macaques intraperitoneally. We repeated the same six conditions of inoculation with PrP-KO mice, as described in Table 1; PrP-KO mice do not develop vCJD, and thus, any PrPTSE detected in their blood must have come from the inoculum, allowing us to determine the kinetics of clearance of PrPTSE remaining from the inoculum after each condition of inoculation.
3.2. PMCA Products Detected Using ELISA-like Assay
We used PMCA to test all blood samples collected during the study, using Western blot to confirm the presence of PMCA products. To detect those products, we employed an ELISA-like assay: a commercial HC PrPTSE test based on a proprietary ligand that selectively binds and concentrates aggregated PrPTSE but not monomeric PrP, requiring no preliminary protease treatment of samples to remove normal PrPC (i.e., no proteinase K digestion) as required to detect PrPTSE by Western blot. We directly compared the sensitivity of the HC PrPTSE test to that of the Western blot (Figure 1) using 2-fold serial dilutions of a PMCA product. PrPTSE signal was detected by Western blot in samples diluted 1:128; the HC PrPTSE test detected PrPTSE only in samples diluted ≤ 1:8, the lowest dilution at which PrPTSE was consistently detected above a threshold value of 0.31 O.D. at 562 nm [27]. This value was established as 3 times the standard deviation of average O.D. values of negative human brain homogenates [27].
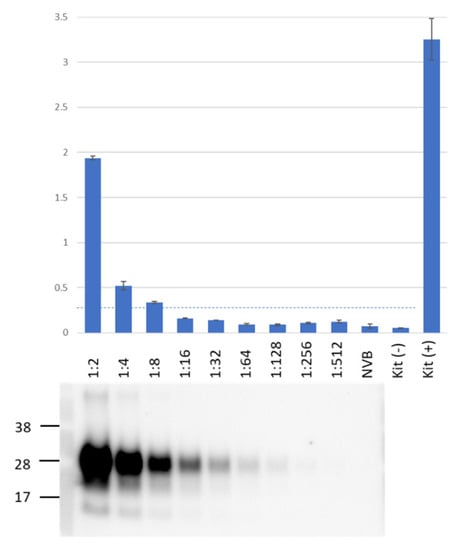
Figure 1.
Direct comparison of assay sensitivity for HerdChek (HC) PrPTSE test and Western blot. PMCA products were serially diluted 2-fold in 10% normal vole brain. We used 25 µL of each sample for HC PrPTSE tests. After proteinase K digestion, the equivalent of 10 µL of each sample was assayed by Western blot. The top panel also shows colorimetric results for each sample and for internal controls (negative and positive) included in the HC kit. The horizontal dotted line corresponds to the assay threshold (O.D. 0.31) [28]. The Western blot lanes were aligned with corresponding HC samples. NVB, normal vole brain (PMCA substrate). Representative results from a total of three independent experiments.
These results contrast with our previous data showing that the HC PrPTSE test detected PrPTSE in human sCJD brain homogenates with approximately 30-fold higher sensitivity than that of Western blot [28]. This finding might be explained if the HC PrPTSE test detected human PrPTSE better than vole PrPTSE (PMCA products in this study were generated using vole brain as the normal PrP substrate) or if PrPTSE in our PMCA products had a conformation that did not bind to the ligand as well as natural PrPTSE, reducing its detection by HC test. Despite this limitation, the HC PrPTSE test was useful to compare PMCA signals from the blood and tissues of inoculated mice.
3.3. PrPTSE in the Blood of PrP-KO Mice
Figure 2 shows HC PrPTSE test results after PMCA of blood from PrP-KO mice inoculated under the six experimental conditions (Table 1). The first two columns model the inoculation conditions used to infect cynomolgus and rhesus macaques.
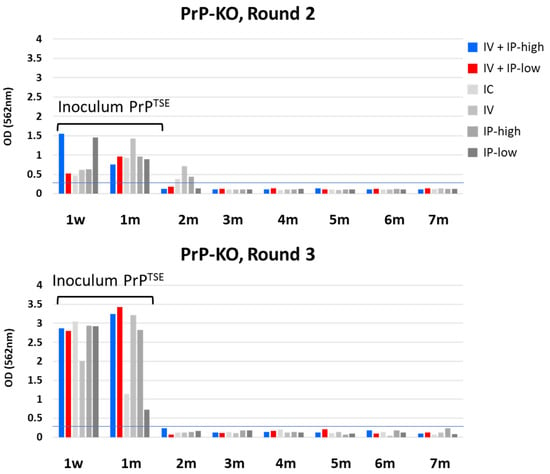
Figure 2.
HC PrPTSE test results for PrP-KO mice. Six groups of PrP-KO mice were inoculated as indicated in the figure. Importantly, “IV + IP-high” matched inoculation conditions used to inoculate cynomolgus macaques, and “IV + IP-low” matched conditions used to inoculate rhesus macaques [18]. Blood aliquots from inoculated mice collected at the indicated times were tested for PrPTSE by PMCA. PMCA products were assayed by HC PrPTSE test, and results for rounds 2 (top panel) and 3 (lower panel) are reported as the colorimetric signal versus collection time. w, week; m, month; IV, intravenous; IP, intraperitoneal; IC, intracerebral.
We detected PMCA products at 1 week and 1 mpi in rounds 2 and 3 of PMCA in blood samples from mice inoculated under all conditions (Figure 2). As expected, the overall levels of PMCA products were higher after round 3 than after round 2 because concentrations of PrPTSE increased with each successive cycle of sonication. PrPTSE was also detected above the threshold in some blood samples after round 2, but no signals were present in PMCA products collected after round 3. Upon close review of Western blots, we detected no PrPTSE in those same samples after PMCA rounds 2 and 3 (Figure 3: PrP-KO blots in panels A and B). Based on these observations, we concluded that the reactive signals detected 2 mpi were probably artifacts of the HC PrPTSE test; we do not know the origin of these apparent false positive signals.
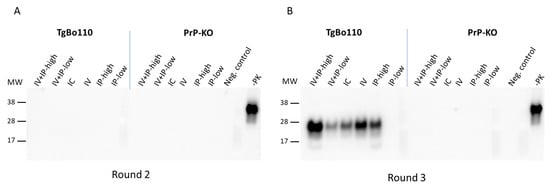
Figure 3.
Comparison of blood of vCJD-inoculated TgBo110 and PrP-KO mice collected 2 months post-inoculation after rounds 2 and 3 of PMCA. Blood aliquots of TgBo110 and PrP-KO mice inoculated as indicated in the figure were assayed for PrPTSE using PMCA. PMCA products were visualized after rounds 2 (panel (A)) and 3 rounds (panel (B)), after proteinase K digestion, on Western blots using antibodies against PrP. “Neg. control”, normal vole brain substrate after PMCA and treatment with proteinase K; “-PK”, normal vole brain without PMCA and not treated with proteinase K; MW, molecular weight markers.
3.4. PrPTSE in the Blood of TgBo110 Mice
We detected PrPTSE remaining from the inoculum in the blood of TgBo110 mice (Figure 4) up to 1 mpi, a finding consistent with those seen with PrP-KO mice (Figure 2). As previously noted, we observed weak signals by HC testing in blood samples from some mice 2 mpi, not confirmed to be PrPTSE by Western blot (Figure 3: TgBo110 blot panel A).
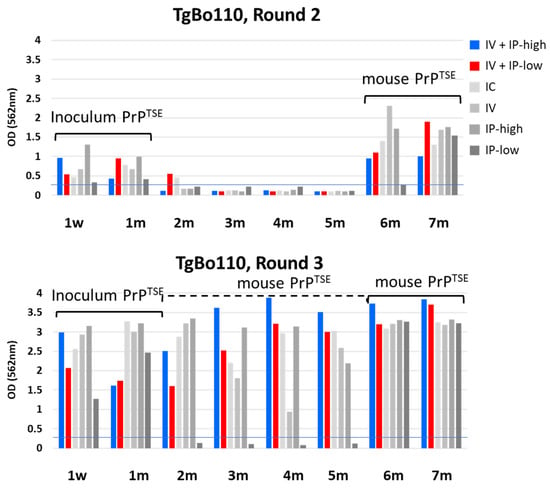
Figure 4.
HC PrPTSE test results for TgBo110 mice. Six groups of TgBo110 mice were inoculated as indicated in the figure. Importantly, the “IV + IP-high” condition matched inoculation conditions for cynomolgus macaques, and “IV + IP-low” matched inoculation conditions for rhesus macaques, as previously reported [18]. Blood aliquots from inoculated mice collected at the indicated times were tested for PrPTSE by PMCA. PMCA products were assayed by HC PrPTSE test, and results for rounds 2 (top panel) and 3 (lower panel) are reported as the colorimetric signal versus collection time. w, week; m, month; IV, intravenous; IP, intraperitoneal; IC, intracerebral.
Once PrPTSE from the inoculum had cleared, no PrPTSE was detected by PMCA during the next 4 months. PrPTSE again appeared 6 mpi in the blood of mice inoculated with vCJD agent under all conditions except not in blood from mice inoculated under IP-low conditions. Seven months post-inoculation, similar amounts of PrPTSE amplified by PMCA were detected in the blood of mice inoculated under all conditions. We conclude that this second wave of PrPTSE detected by PMCA must have been generated de novo by infected mice. Interestingly, round 3 of PMCA detected PrPTSE even in samples negative in round 2. We confirmed these results by Western blot (Figure 3B, left panel) showing PrPTSE in the blood of mice inoculated under all conditions except for the IP-low condition. These observations indicated that de novo mouse-generated PrPTSE was already present in the blood of TgBo110 mice as early as 2 to 5 mpi, but only in very low concentrations—undetectable by the first two rounds of PMCA. It is also important to note that the results for mice inoculated under two experimental conditions mimicking those previously used with cynomolgus and rhesus macaques showed similar results. These data suggest that the difference in dose of inoculum did not affect the time of appearance of PrPTSE in blood.
3.5. Final Disease Status of Each Mouse in the Study
We monitored TgBo110 mice until the end of the study and confirmed that de novo-generated PrPTSE remained detectable in their blood from its first appearance until terminal illness. Table 2 shows a complete analysis of TgBo110 mice with results from PMCA tests of blood, brain, and spleen for each animal. All mice, except five, at the time of death, had evidence of infection confirmed by PMCA reactivity in at least one tissue tested: blood, brain, and spleen. Of the five mice with negative results, two died too early to know whether they were infected; the brains of two other mice were negative, but their blood and spleen—earlier indicators of infection—could not be tested. One mouse in the IP-low cohort was truly uninfected because it died late in the vCJD incubation period and the PMCA of all three tissues was negative. Scheduled euthanasia was conducted 4 mpi (for one mouse 3 mpi) to determine the status of TgBo110 mice at early time points when we expected animals to have no indications of vCJD. In tissues of mice inoculated under all conditions except IC inoculations, blood, and spleen were PMCA-positive before PrPTSE was detected in the brain. To our surprise, PrPTSE was already detectable in the blood (and spleen) as early as 4 mpi except for IP-low conditions. It is also noteworthy that PrPTSE was detectable in the brain no earlier than 12 mpi while blood was PrPTSE-positive already at 3 or 4 mpi for mice inoculated under all conditions except for mice inoculated under the IP-low condition. These results confirm that, regardless of the inoculation route, blood and spleen are affected very early in the infection. When we compared mice inoculated under conditions intended to model previous cynomolgus and rhesus inoculations, we observed that the blood of both cohorts of mice became PrPTSE-positive at the same times, as early as 3 or 4 mpi.
Table 2.
Summary of PrPTSE detection for each inoculated mouse.
4. Discussion
PrPTSE is currently the only biomarker specific for TSE diseases. However, we know very little about this protein, especially regarding the properties of PrPTSE in blood: its biochemical characteristics, its time of appearance in preclinical phases of infection and clinical illness, and changes in its concentration during the course of infection and from individual to individual. These are important questions to address before accepting PrPTSE as a reliable biomarker suitable to develop into useful antemortem diagnostic assays for human TSEs. Studying PrPTSE in blood poses challenges, the first being due to its very low concentration requiring in vitro PrPTSE amplification tests such as PMCA or RT-QuIC for its detection and the second being the lack of blood samples collected from infected humans spanning the period from preclinical “incubation period” to overt clinical illness. To overcome the second limitation, we and other investigators experimentally infected large TSE-susceptible animals and collected their blood throughout the incubation period for testing to detect PrPTSE. For example, PrPTSE was first detected in the blood of transfused sheep 6 months post-exposure, the earliest time point tested, and then found continuously in blood samples collected before clinical onset and during overt illness until euthanasia [29]. In another study, longitudinal blood samples from macaques infected intravenously with brain-derived vCJD agent tested PrPTSE-positive by PMCA as early as 10 mpi [12]. RT-QuIC detected PrPTSE in the blood of deer 1–2 days post-inoculation, after which the signal declined but subsequently increased sharply at 3 mpi, remaining constant for several months through the end of the study [30]. Those results are consistent with the findings of Soto and colleagues, and our own, using blood of macaques experimentally infected with vCJD by the IV and IP routes [18,31]. Collectively, these studies revealed that PrPTSE could be detected in blood relatively soon after exposure and for several months before clinical onset. However, these studies left open the possibility that the PrPTSE detected early was a residuum from the inoculum remaining in the blood. This option could not be excluded because the in vitro amplification assays did not distinguish PrPTSE from the brain (the inoculum) or blood (generated by infected animals de novo). We used mice to investigate this question. Compared to nonhuman primates, mice have the advantage of being available as PrP-KO animals, unable to be infected with TSEs or to generate endogenous PrPTSE, allowing us to follow the fate of PrPTSE remaining from the inoculum. Mice are also ideal for comparative studies using different routes of inoculation, research requiring an unfeasible number of nonhuman primates. Mice are inbred and homogeneous, unlike macaques which are outbred and more variable in genome and phenotype. Caveats regarding mouse studies are that PrPTSE in their blood might be cleared by mechanisms that differ from those of primates, yielding differences in kinetics. Blood samples must also be pooled from several mice in the same cohort to obtain sufficient volumes for testing. Pooling blood samples loses the opportunity to investigate the variations from animal to animal that we observed with our studies in macaques.
Studies with PrP-KO mice informed us that PrPTSE from the inoculum remained detectable up to 1 month post-exposure but was completely cleared by 2 mpi. These results were consistent for all conditions of inoculation tested. The same clearance of PrPTSE originating from the inoculum was also observed with TgBo110 mice and followed by a period of 4 months during which extremely low levels of PrPTSE in the blood required three rounds of PMCA to detect. PrPTSE then increased dramatically in blood 6 mpi and remained constant throughout the rest of the incubation period and during overt vCJD. Because PrPTSE was not detected in the blood of PrP-KO mice 2 mpi even by round 3 of PMCA, we can be confident that PrPTSE was generated de novo by vCJD-infected TgBo110 mice. Mice infected under all conditions of inoculation showed the same trend of PrPTSE detected in blood. Although PrPTSE clearance in mice might be different than in macaques, the complete clearance of inoculated PrPTSE from the blood of our PrP-KO mice during the first two months after inoculations suggests that the same could have occurred in infected macaques. If this is correct, PrPTSE was released into the blood of infected cynomolgus macaques more than 2 years before they showed clinical signs of vCJD. This conclusion is consistent with reported cases of transfusion-transmitted vCJD in recipients of blood donated 18 to 40 months before donors became clinically ill with vCJD [6]. Our results are also in agreement with the detection of PrPTSE in the blood of two vCJD-infected asymptomatic individuals 14 and 31 months prior to the earliest clinical signs of illness [14]. Taken together, these and our findings are encouraging for prospects to develop informative assays that detect PrPTSE in the blood of infected people before the clinical onset of illness. In addition, our observations highlight the importance of beginning the collection of blood samples early when testing for PrPTSE in future experimental studies of TSEs in large animals.
We showed that PrPTSE from the inoculum remained circulating in the blood of inoculated mice for at least 1 month, a time that differs from a half-life of only a few hours estimated by a study with purified radiolabeled and non-radiolabeled PrPTSE inoculated intravenously into mice [32]. There could be several reasons for these different results. An example is differences in the preparation of inocula: in our studies, PrPTSE was in crude brain homogenate preparations also containing a variety of proteins, lipids, and other components that might protect PrPTSE from degradation or retard its uptake into tissues. In contrast, radiolabeled studies used highly purified and chemically modified PrPTSE. Furthermore, we injected more PrPTSE, which may have prolonged the time PrPTSE circulated in blood. Importantly, Urayama and colleagues, in the same studies, showed rapid uptake of IV-injected PrPTSE by the spleen and other lymphoid tissues, sites likely to participate in the peripheral replication of infectivity and generation of PrPTSE [32]. Consistent with their findings, we detected PrPTSE in spleens as early as 3 mpi (earliest time point tested)—much earlier than in the brain. Considering its probable early involvement in the development of vCJD after peripheral routes of infection, the spleen might be a common site where murine PrPTSE is generated de novo and released into the circulation. Further studies would be needed to confirm this hypothesis.
Our mouse studies indicated no difference in the times when endogenous PrPTSE was first detected in the blood of mice inoculated with vCJD agent under conditions that mimicked those used to infect cynomolgus and rhesus macaques. This suggested that the reduced inoculum dose used to infect rhesus macaques was unlikely to have caused the delayed appearance of PrPTSE in rhesus blood. We hypothesized that some weak species barrier in rhesus macaques inoculated with cynomolgus-derived inoculum might have allowed vCJD to transmit more efficiently to cynomolgus than to rhesus macaques—an unexpected finding since both species of macaque are genetically very close. Alternatively, natural variability from animal to animal might also explain those differences. In any case, these findings serve as a warning that tests that detect PrPTSE in blood might not always be reliable, reducing their negative predictive value.
In conclusion, independent of the route of experimental exposure to the vCJD agent, PrPTSE was detected in the blood of susceptible mice starting a few months post-exposure and maintained at constant levels in the blood for years. These data support efforts to develop and validate tests for PrPTSE in blood to identify individuals with early TSE infections and, thus, reduce the risk of iatrogenic sCJD. We also detected PrPTSE products amplified from mouse blood by PMCA using a PrPTSE ELISA-like assay that was rapid and reasonably sensitive, with a readout simpler, faster, and less variable than that of Western blots. Improvement of high-throughput ELISA-like assays to detect human PrPTSE might eventually facilitate the development of a human TSE test.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/v15071466/s1, Figure S1: Experimental design.
Author Contributions
Conceptualization, O.Y. and L.G.; methodology, O.Y. and T.P.; writing—original draft preparation, L.G.; writing—review and editing, O.Y., T.P., D.M.A. and L.G.; supervision, L.G. and D.M.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the United States Food and Drug Administration. The funders of the work did not influence the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional Review Board Statement
Animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the U.S. National Institutes of Health (8th Ed.) and requirements of US Animal Welfare Regulations. The protocols were reviewed and approved by the Institutional Animal Care and Use Committee at FDA (ASP2010-05). The FDA CBER-managed animal program and facilities are fully accredited by AAALAC International.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the technical staff of CBER’s Division of Veterinary Services for their dedicated and excellent care of the animals. We are particularly indebted to Jaroslav Vostal and colleagues at FDA for generously sharing breeding pairs of PrP-knockout mice. We also thank members of our laboratory for their support of this project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bonda, D.J.; Manjila, S.; Mehndiratta, P.; Khan, F.; Miller, B.R.; Onwuzulike, K.; Puoti, G.; Cohen, M.L.; Schonberger, L.B.; Cali, I. Human prion diseases: Surgical lessons learned from iatrogenic prion transmission. Neurosurg. Focus 2016, 41, E10. [Google Scholar] [CrossRef] [PubMed]
- Ironside, J.W. Variant Creutzfeldt-Jakob disease: An update. Folia Neuropathol. 2012, 50, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Will, R. Variant Creutzfeldt-Jakob disease. Folia Neuropathol. 2004, 42 (Suppl. A), 77–83. [Google Scholar] [PubMed]
- Belay, E.D.; Schonberger, L.B. Variant Creutzfeldt-Jakob disease and bovine spongiform encephalopathy. Clin. Lab. Med. 2002, 22, 849–862. [Google Scholar] [CrossRef]
- Watson, N.; Brandel, J.P.; Green, A.; Hermann, P.; Ladogana, A.; Lindsay, T.; Mackenzie, J.; Pocchiari, M.; Smith, C.; Zerr, I.; et al. The importance of ongoing international surveillance for Creutzfeldt-Jakob disease. Nat. Rev. Neurol. 2021, 17, 362–379. [Google Scholar] [CrossRef]
- Ritchie, D.L.; Peden, A.H.; Barria, M.A. Variant CJD: Reflections a Quarter of a Century on. Pathogens 2021, 10, 1413. [Google Scholar] [CrossRef]
- Hermann, P.; Appleby, B.; Brandel, J.P.; Caughey, B.; Collins, S.; Geschwind, M.D.; Green, A.; Haïk, S.; Kovacs, G.G.; Ladogana, A.; et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol. 2021, 20, 235–246. [Google Scholar] [CrossRef]
- Schmitz, M.; Dittmar, K.; Llorens, F.; Gelpi, E.; Ferrer, I.; Schulz-Schaeffer, W.J.; Zerr, I. Hereditary human prion diseases: An update. Mol. Neurobiol. 2017, 54, 4138–4149. [Google Scholar] [CrossRef]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef]
- Atarashi, R.; Wilham, J.M.; Christensen, L.; Hughson, A.G.; Moore, R.A.; Johnson, L.M.; Onwubiko, H.A.; Priola, S.A.; Caughey, B. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat. Methods 2008, 5, 211–212. [Google Scholar] [CrossRef]
- Green, A.J.E.; Zanusso, G. Prion protein amplification techniques. Handb. Clin. Neurol. 2018, 153, 357–370. [Google Scholar]
- Lacroux, C.; Comoy, E.; Moudjou, M.; Perret-Liaudet, A.; Lugan, S.; Litaise, C.; Simmons, H.; Jas-Duval, C.; Lantier, I.; Beringue, V.; et al. Preclinical detection of variant CJD and BSE prions in blood. PLoS Pathog. 2014, 10, e1004202. [Google Scholar] [CrossRef]
- Concha-Marambio, L.; Pritzkow, S.; Moda, F.; Tagliavini, F.; Ironside, J.W.; Schulz, P.E.; Soto, C. Detection of prions in blood from patients with variant Creutzfeldt-Jakob disease. Sci. Transl. Med. 2016, 8, 370ra183. [Google Scholar] [CrossRef] [PubMed]
- Bougard, D.; Brandel, J.P.; Bélondrade, M.; Béringue, V.; Segarra, C.; Fleury, H.; Laplanche, J.L.; Mayran, C.; Nicot, S.; Green, A.; et al. Detection of prions in the plasma of presymptomatic and symptomatic patients with variant Creutzfeldt-Jakob disease. Sci. Transl. Med. 2016, 8, 370ra182. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.S.; Burk-Rafel, J.; Edgeworth, J.A.; Sicilia, A.; Abdilahi, S.; Korteweg, J.; Mackey, J.; Thomas, C.; Wang, G.; Mead, S.; et al. A highly specific blood test for vCJD. Blood 2014, 123, 452–453. [Google Scholar] [CrossRef] [PubMed]
- Douet, J.Y.; Zafar, S.; Perret-Liaudet, A.; Lacroux, C.; Lugan, S.; Aron, N.; Cassard, H.; Ponto, C.; Corbière, F.; Torres, J.M.; et al. Detection of infectivity in blood of persons with variant and sporadic Creutzfeldt-Jakob disease. Emerg. Infect. Dis. 2014, 20, 114–117. [Google Scholar] [CrossRef]
- McDowell, K.L.; Nag, N.; Franco, Z.; Bu, M.; Piccardo, P.; Cervenak, J.; Deslys, J.P.; Comoy, E.; Asher, D.M.; Gregori, L. Blood reference materials from macaques infected with variant Creutzfeldt-Jakob disease agent. Transfusion 2015, 55, 405–412. [Google Scholar] [CrossRef]
- Yakovleva, O.; Bett, C.; Pilant, T.; Asher, D.M.; Gregori, L. Abnormal prion protein, infectivity and neurofilament light-chain in blood of macaques with experimental variant Creutzfeldt-Jakob disease. J. Gen. Virol. 2022, 103, 001764. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Gutiérrez Adán, A.; Brun, A.; Pintado, B.; Ramírez, M.A.; Parra, B.; Doyle, D.; Rogers, M.; Salguero, F.J.; Sánchez, C.; et al. Early detection of PrPres in BSE-infected bovine PrP transgenic mice. Arch. Virol. 2003, 148, 677–691. [Google Scholar]
- Bett, C.; Piccardo, P.; Cervenak, J.; Torres, J.M.; Asher, D.M.; Gregori, L. Both murine host and inoculum modulate expression of experimental variant Creutzfeldt-Jakob disease. J. Gen. Virol. 2018, 99, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Serra, F.; Dudas, S.; Torres, J.M.; Anderson, R.; Oevermann, A.; Espinosa, J.C.; Czub, S.; Seuberlich, T. Presumptive BSE cases with an aberrant prion protein phenotype in Switzerland, 2011: Lack of prion disease in experimentally inoculated cattle and bovine prion protein transgenic mice. Transbound Emerg. Dis. 2018, 65, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Büeler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, E.B.; Edgeworth, J.A.; Thomas, C.; Collinge, J.; Jackson, G.S. Preclinical detection of infectivity and disease-specific PrP in blood throughout the incubation period of prion disease. Sci. Rep. 2015, 5, 17742. [Google Scholar] [CrossRef]
- Saá, P.; Castilla, J.; Soto, C. Presymptomatic detection of prions in blood. Science 2006, 313, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Glier, H.; Simak, J.; Panigaj, M.; Gelderman, M.P.; Vostal, J.G.; Holada, K. Expression of the cellular prion protein affects posttransfusion recovery and survival of red blood cells in mice. Transfusion 2015, 55, 2590–2596. [Google Scholar] [CrossRef]
- Diehl, K.H.; Hull, R.; Morton, D.; Pfister, R.; Rabemampianina, Y.; Smith, D.; Vidal, J.M.; Vorstenbosch, C.V.D. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J. Appl. Toxicol. 2001, 21, 15–23. [Google Scholar] [CrossRef]
- Nemecek, J.; Nag, N.; Carlson, C.M.; Schneider, J.R.; Heisey, D.M.; Johnson, C.J.; Asher, D.M.; Gregori, L. Red-backed vole brain promotes highly efficient in vitro amplification of abnormal prion protein from macaque and human brains infected with variant Creutzfeldt-Jakob disease agent. PLoS ONE 2013, 8, e78710. [Google Scholar] [CrossRef]
- Gregori, L.; Serer, A.R.; McDowell, K.L.; Cervenak, J.; Asher, D.M. Rapid Testing for Creutzfeldt-Jakob disease in donors of cornea. Transplantation 2017, 101, e120–e124. [Google Scholar] [CrossRef]
- Salamat, M.K.F.; Stewart, P.; Brown, H.; Tan, K.B.C.; Smith, A.; de Wolf, C.; Alejo Blanco, A.R.; Turner, M.; Manson, J.C.; McCutcheon, S.; et al. Subclinical infection occurs frequently following low dose exposure to prions by blood transfusion. Sci. Rep. 2022, 12, 10923. [Google Scholar] [CrossRef]
- Elder, A.M.; Henderson, D.M.; Nalls, A.V.; Hoover, E.A.; Kincaid, A.E.; Bartz, J.C.; Mathiason, C.K. Immediate and ongoing detection of prions in the blood of hamsters and deer following oral, nasal, or blood inoculations. J. Virol. 2015, 89, 7421–7424. [Google Scholar] [CrossRef]
- Concha-Marambio, L.; Chacon, M.A.; Soto, C. Preclinical detection of prions in blood of nonhuman primates infected with variant Creutzfeldt-Jakob disease. Emerg. Infect. Dis. 2020, 26, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Urayama, A.; Morales, R.; Niehoff, M.L.; Banks, W.A.; Soto, C. Initial fate of prions upon peripheral infection: Half-life, distribution, clearance, and tissue uptake. FASEB J. 2011, 25, 2792–2803. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).