
Structural Model for Factor X Inhibition of IgM and Complement-Mediated Neutralization of Adenovirus


Abstract
1. Introduction
2. Materials and Methods
2.1. Initial Model Building
2.2. Modeling Addition of C3b to the Facet
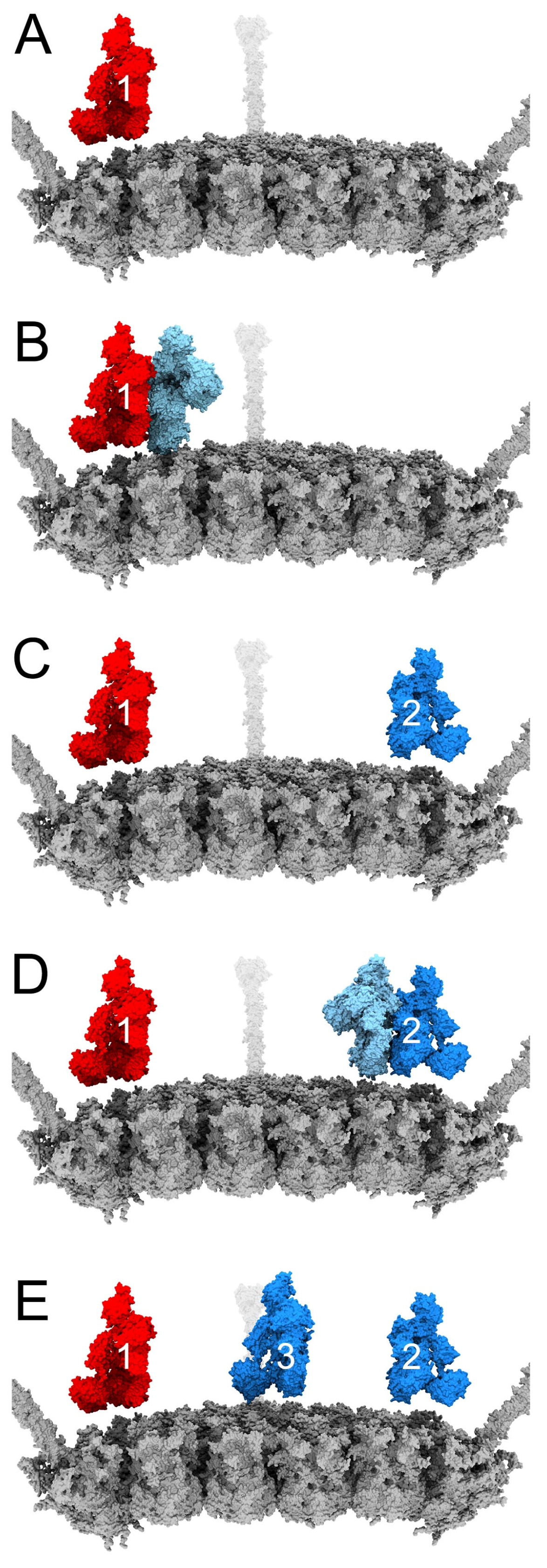
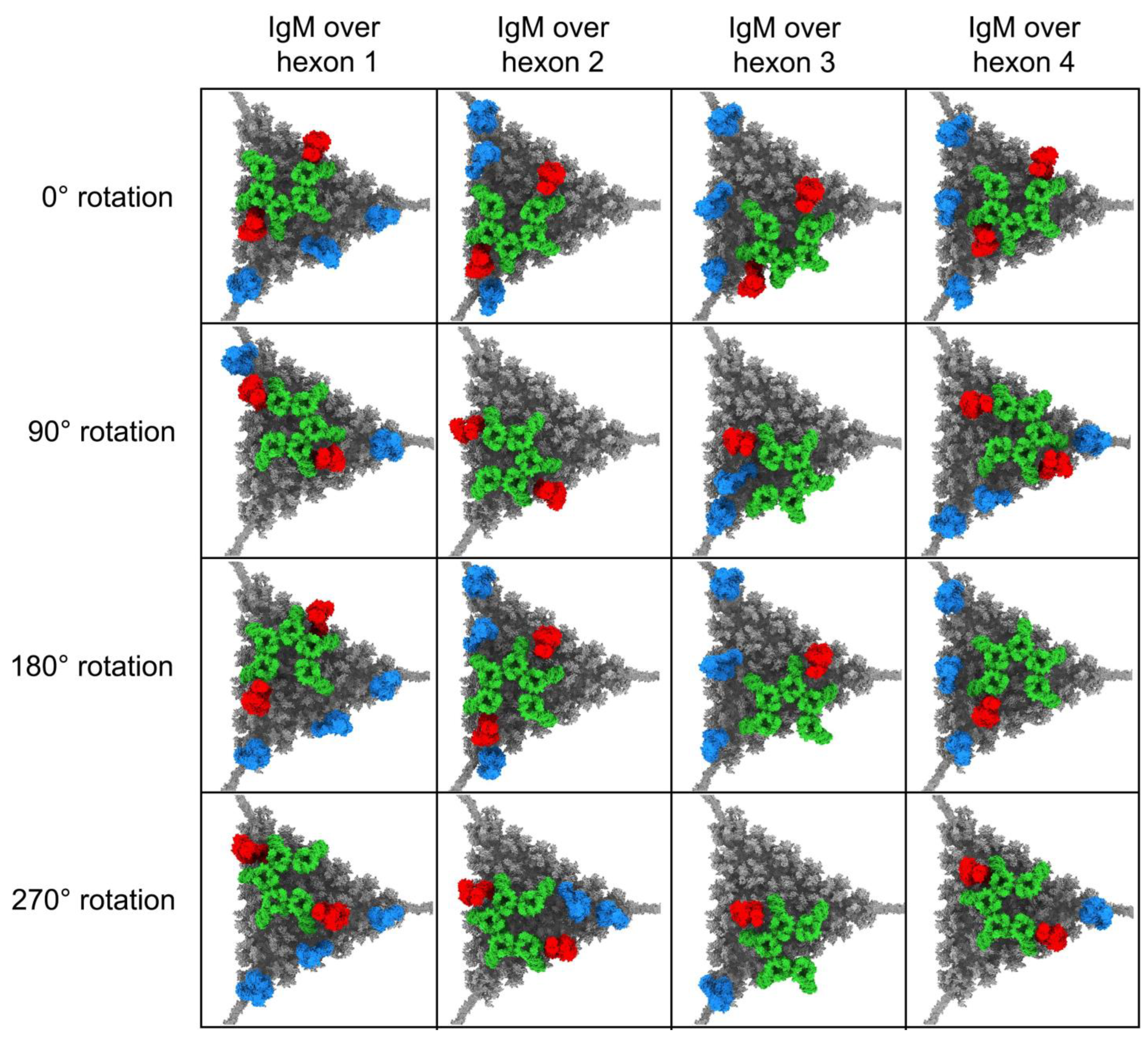
2.3. Modeling Competition between FX and IgM on HAdv-C5
2.4. Molecular Dynamics Simulations
2.5. Calculation of Non-Bonded Interaction Energies
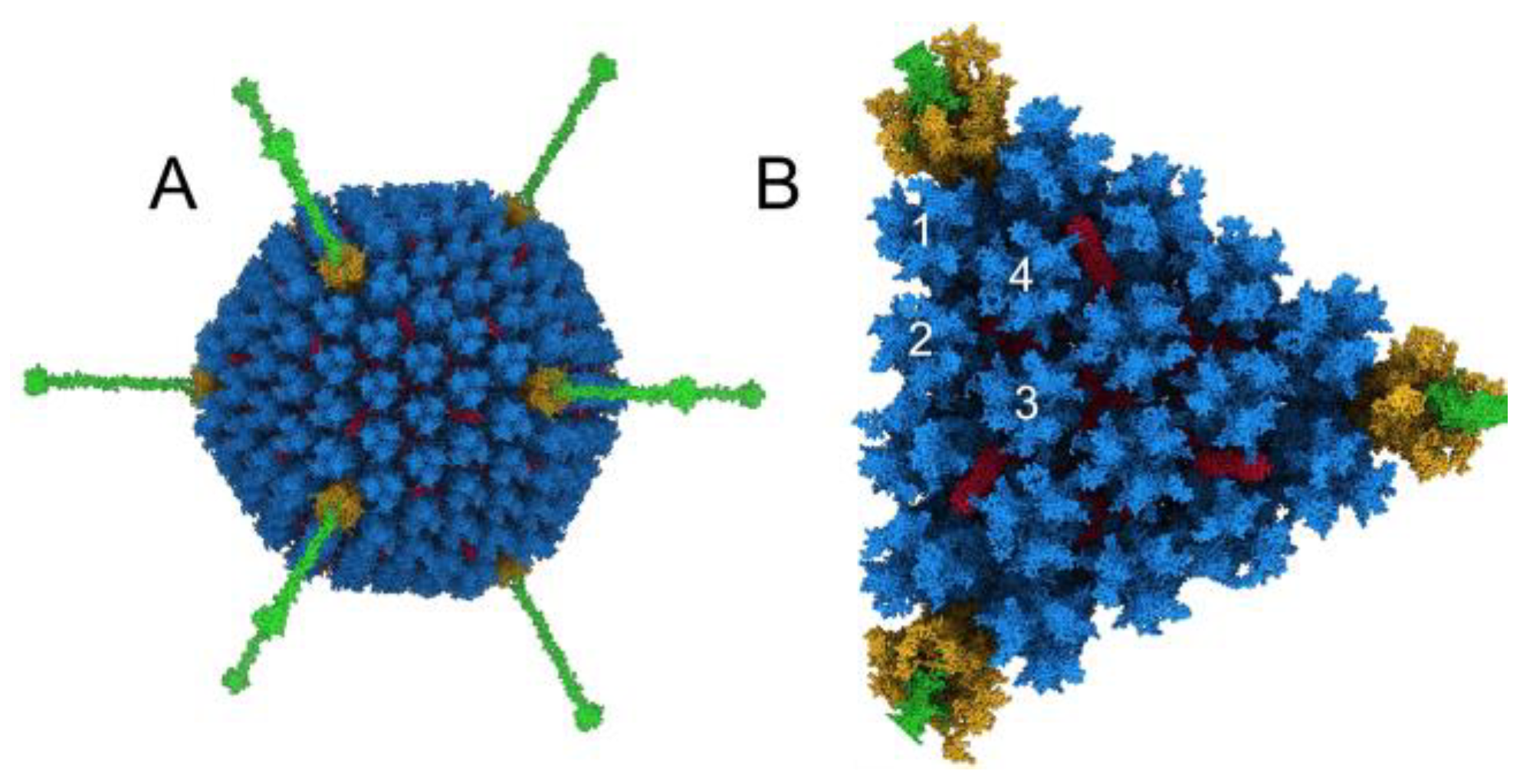
3. Results
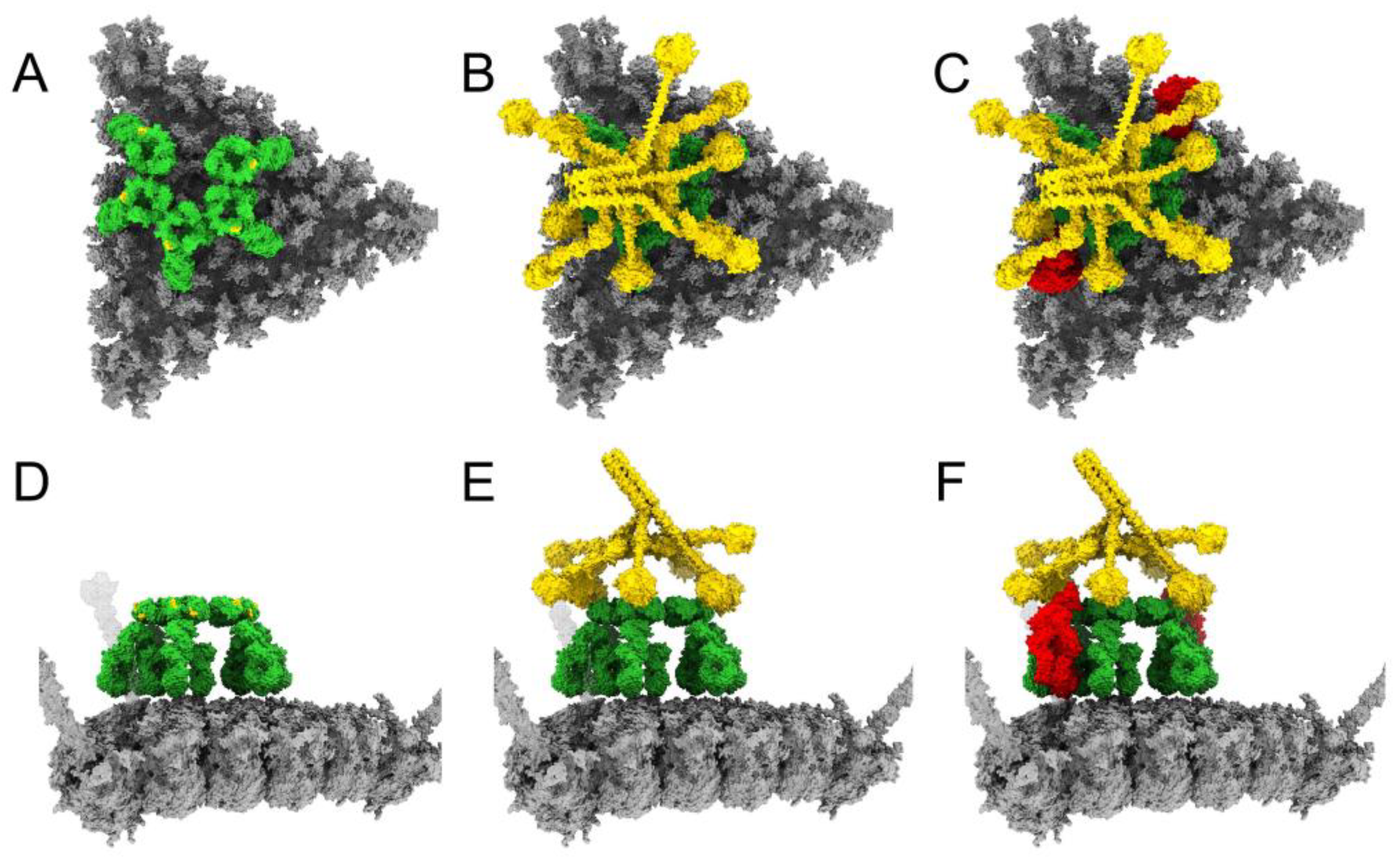
3.1. Modeling Natural IgM, Complement C1, and C4b Binding to HAdv-C5
3.2. Complement C3b Covalent Binding and Amplification
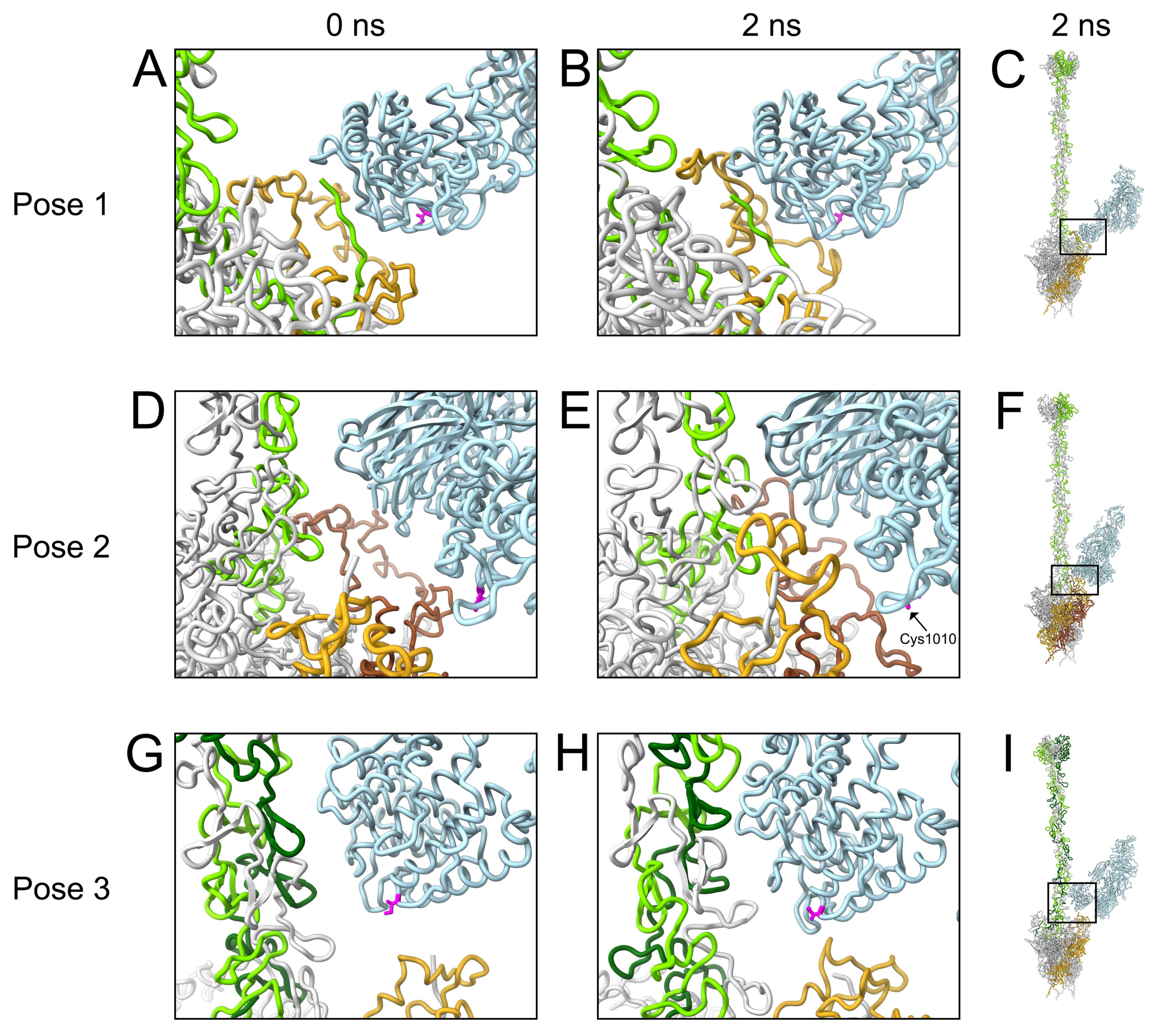
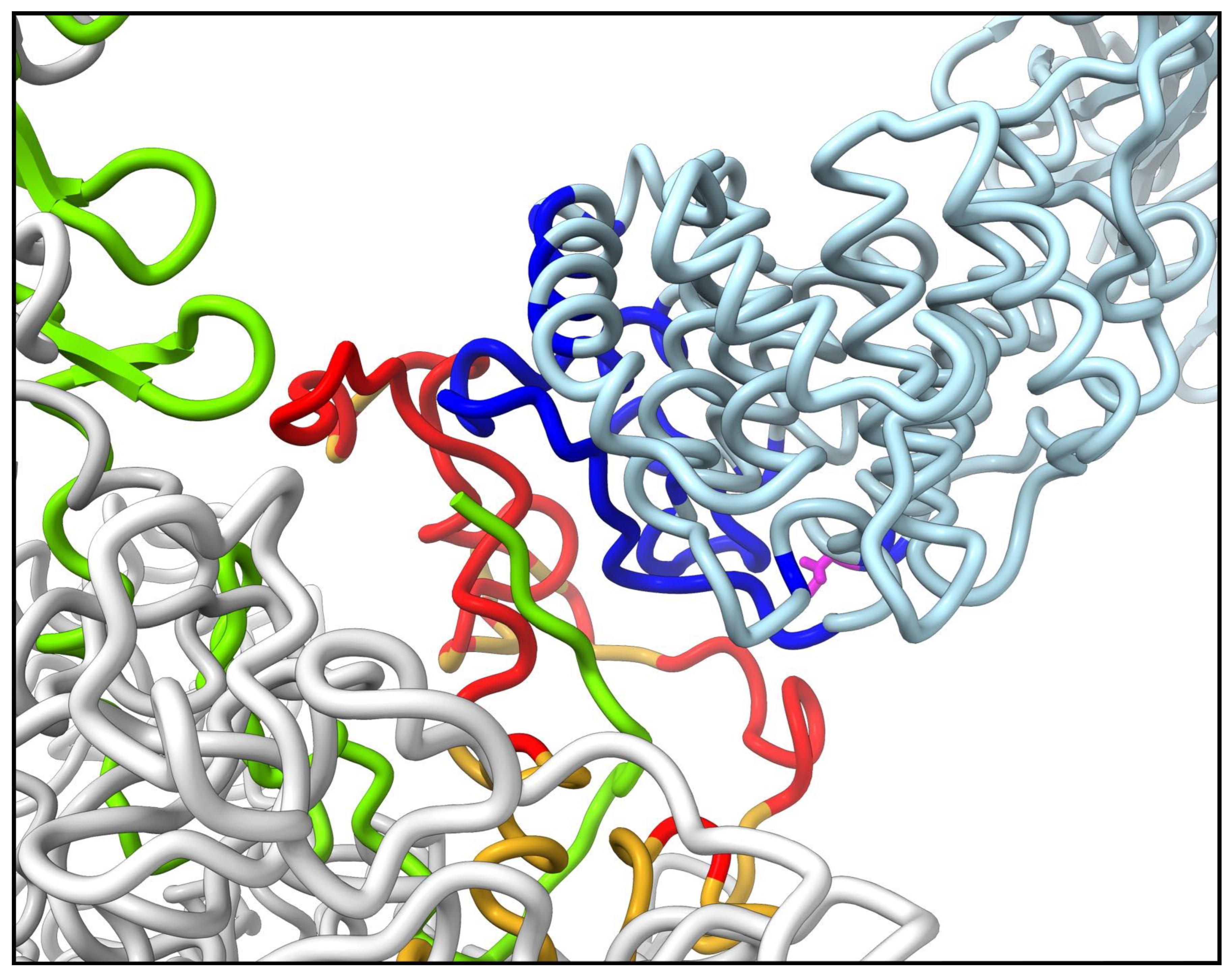
3.3. Molecular Dynamics Simulations with HAdv-C5 Penton Base, Fiber, and C3b
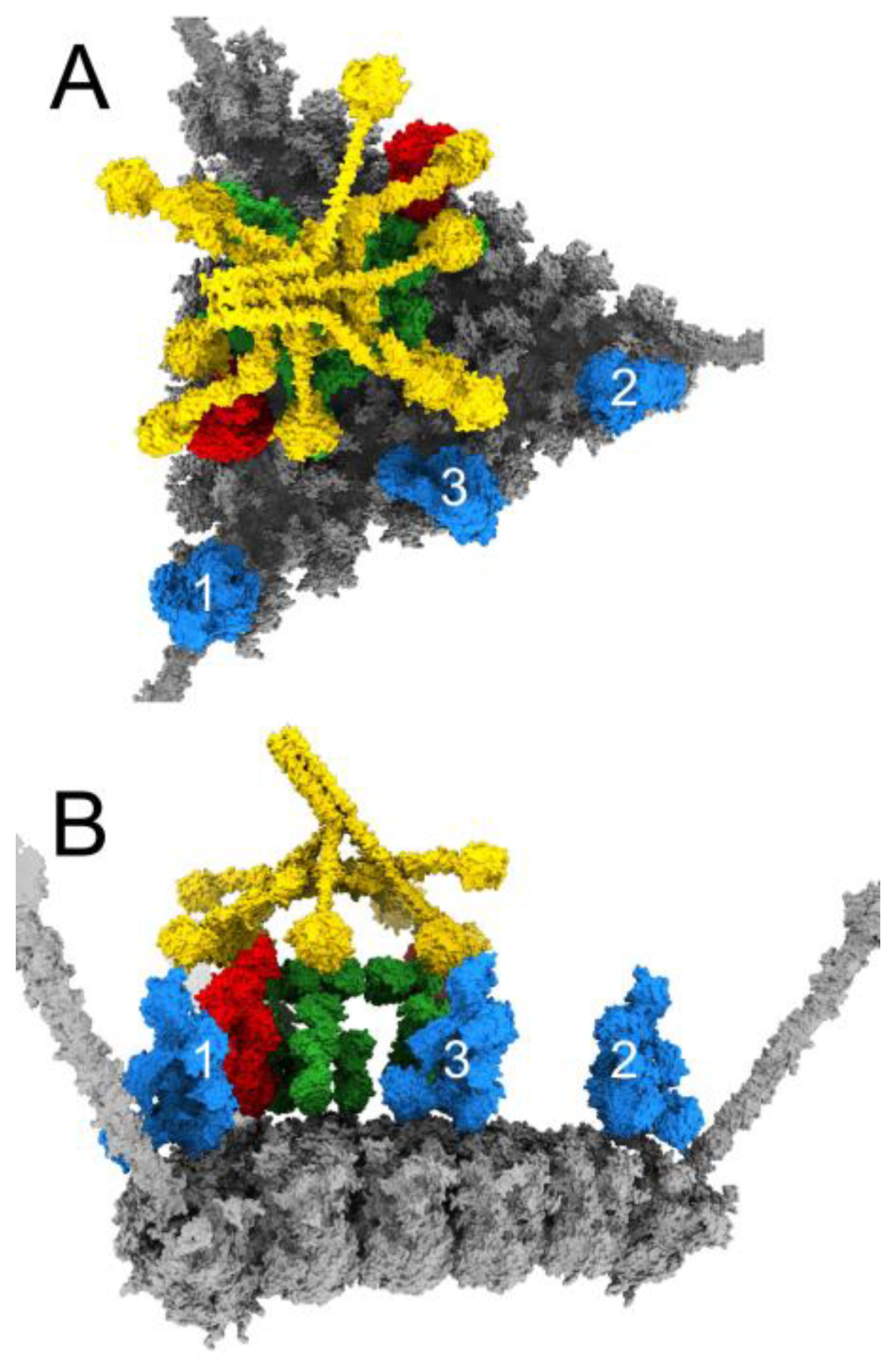
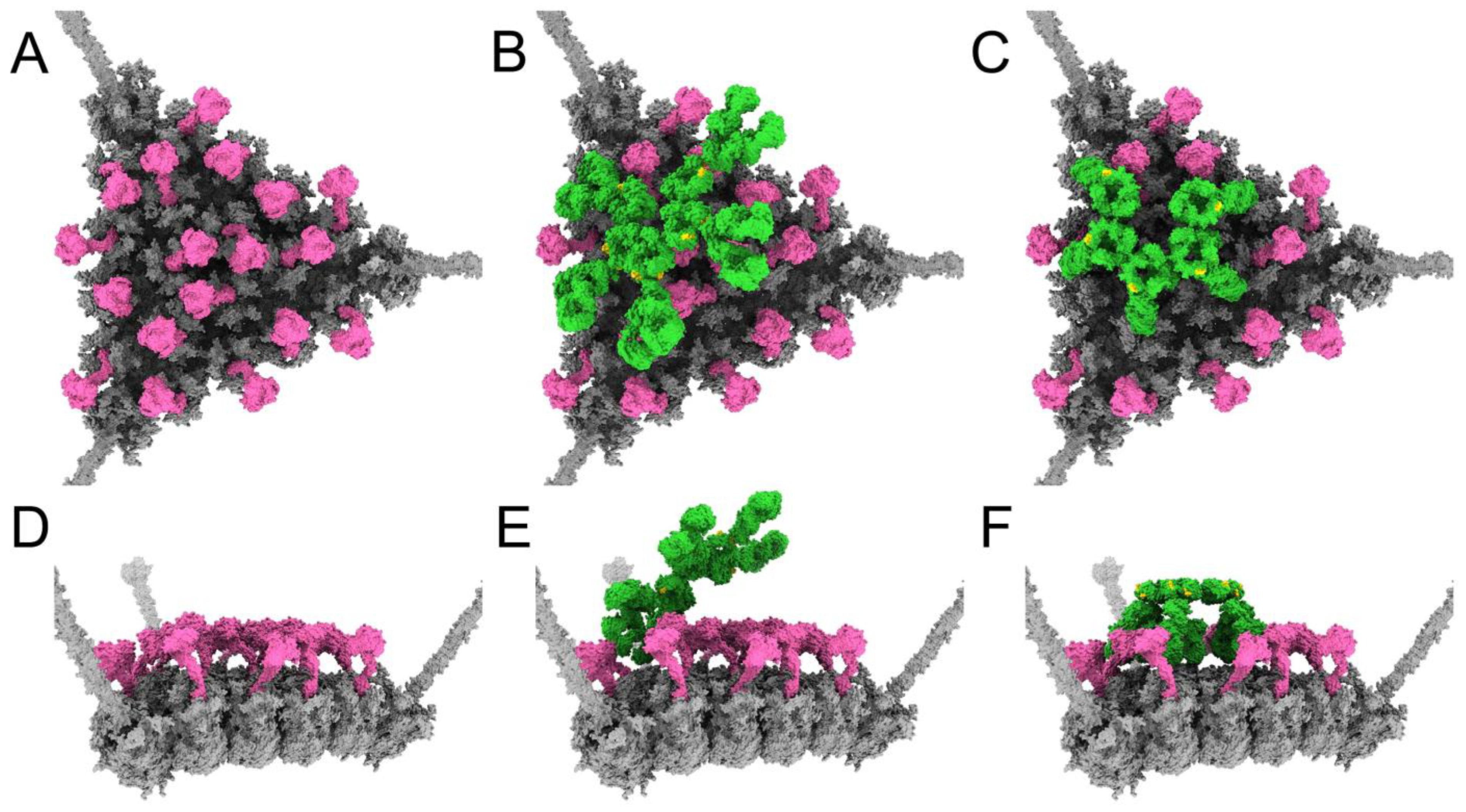
3.4. Structural Model for Factor X Inhibition of IgM and Complement-Mediated Neutralization of HAdv-C5
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garcia-Carbonero, R.; Bazan-Peregrino, M.; Gil-Martin, M.; Alvarez, R.; Macarulla, T.; Riesco-Martinez, M.C.; Verdaguer, H.; Guillen-Ponce, C.; Farrera-Sal, M.; Moreno, R.; et al. Phase I, multicenter, open-label study of intravenous VCN-01 oncolytic adenovirus with or without nab-paclitaxel plus gemcitabine in patients with advanced solid tumors. J. Immunother. Cancer 2022, 10, e003255. [Google Scholar] [CrossRef]
- Li, C.; Wang, H.; Georgakopoulou, A.; Gil, S.; Yannaki, E.; Lieber, A. In Vivo HSC Gene Therapy Using a Bi-modular HDAd5/35++ Vector Cures Sickle Cell Disease in a Mouse Model. Mol. Ther. 2021, 29, 822–837. [Google Scholar] [CrossRef]
- O’Cathail, S.M.; Davis, S.; Holmes, J.; Brown, R.; Fisher, K.; Seymour, L.; Adams, R.; Good, J.; Sebag-Montefiore, D.; Maughan, T.; et al. A phase 1 trial of the safety, tolerability and biological effects of intravenous Enadenotucirev, a novel oncolytic virus, in combination with chemoradiotherapy in locally advanced rectal cancer (CEDAR). Radiat. Oncol. 2020, 15, 151. [Google Scholar] [CrossRef]
- Wang, H.; Georgakopoulou, A.; Zhang, W.; Kim, J.; Gil, S.; Ehrhardt, A.; Lieber, A. HDAd6/35++—A new helper-dependent adenovirus vector platform for in vivo transduction of hematopoietic stem cells. Mol. Ther. Methods Clin. Dev. 2023, 29, 213–226. [Google Scholar] [CrossRef]
- Allen, R.J.; Byrnes, A.P. Interaction of adenovirus with antibodies, complement, and coagulation factors. FEBS Lett. 2019, 593, 3449–3460. [Google Scholar] [CrossRef]
- Bottermann, M.; Foss, S.; Caddy, S.L.; Clift, D.; van Tienen, L.M.; Vaysburd, M.; Cruickshank, J.; O’Connell, K.; Clark, J.; Mayes, K.; et al. Complement C4 Prevents Viral Infection through Capsid Inactivation. Cell Host Microbe 2019, 25, 617–629.e7. [Google Scholar] [CrossRef]
- Smith, J.G.; Nemerow, G.R. Complement Seals a Virus to Block Infection. Cell Host Microbe 2019, 25, 482–483. [Google Scholar] [CrossRef]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef]
- Maloney, B.E.; Perera, K.D.; Saunders, D.R.D.; Shadipeni, N.; Fleming, S.D. Interactions of viruses and the humoral innate immune response. Clin. Immunol. 2020, 212, 108351. [Google Scholar] [CrossRef]
- Feinstein, A.; Richardson, N.; Taussig, M.I. Immunoglobulin flexibility in complement activation. Immunol. Today 1986, 7, 169–174. [Google Scholar] [CrossRef]
- Barnum, S.R. Complement: A primer for the coming therapeutic revolution. Pharmacol. Ther. 2017, 172, 63–72. [Google Scholar] [CrossRef]
- Duval, A.; Fremeaux-Bacchi, V. Complement biology for hematologists. Am. J. Hematol. 2023, 98 (Suppl. 4), S5–S19. [Google Scholar] [CrossRef]
- Arya, S.; Chen, F.; Spycher, S.; Isenman, D.E.; Shulman, M.J.; Painter, R.H. Mapping of amino acid residues in the C mu 3 domain of mouse IgM important in macromolecular assembly and complement-dependent cytolysis. J. Immunol. 1994, 152, 1206–1212. [Google Scholar] [CrossRef]
- Sharp, T.H.; Boyle, A.L.; Diebolder, C.A.; Kros, A.; Koster, A.J.; Gros, P. Insights into IgM-mediated complement activation based on in situ structures of IgM-C1-C4b. Proc. Natl. Acad. Sci. USA 2019, 116, 11900–11905. [Google Scholar] [CrossRef]
- Geisbrecht, B.V.; Lambris, J.D.; Gros, P. Complement component C3: A structural perspective and potential therapeutic implications. Semin. Immunol. 2022, 59, 101627. [Google Scholar] [CrossRef]
- Thomas, M.L.; Janatova, J.; Gray, W.R.; Tack, B.F. Third component of human complement: Localization of the internal thiolester bond. Proc. Natl. Acad. Sci. USA 1982, 79, 1054–1058. [Google Scholar] [CrossRef]
- Moore, S.R.; Menon, S.S.; Cortes, C.; Ferreira, V.P. Hijacking Factor H for Complement Immune Evasion. Front. Immunol. 2021, 12, 602277. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Gaggar, A.; Ni, S.; Li, Z.Y.; Lieber, A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J. Virol. 2005, 79, 7478–7491. [Google Scholar] [CrossRef]
- Doronin, K.; Flatt, J.W.; Di Paolo, N.C.; Khare, R.; Kalyuzhniy, O.; Acchione, M.; Sumida, J.P.; Ohto, U.; Shimizu, T.; Akashi-Takamura, S.; et al. Coagulation factor X activates innate immunity to human species C adenovirus. Science 2012, 338, 795–798. [Google Scholar] [CrossRef]
- Kalyuzhniy, O.; Di Paolo, N.C.; Silvestry, M.; Hofherr, S.E.; Barry, M.A.; Stewart, P.L.; Shayakhmetov, D.M. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 5483–5488. [Google Scholar] [CrossRef]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.; Greig, J.A.; Denby, L.; et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef]
- Xu, Z.; Qiu, Q.; Tian, J.; Smith, J.S.; Conenello, G.M.; Morita, T.; Byrnes, A.P. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat. Med. 2013, 19, 452–457. [Google Scholar] [CrossRef]
- Duffy, M.R.; Doszpoly, A.; Turner, G.; Nicklin, S.A.; Baker, A.H. The relevance of coagulation factor X protection of adenoviruses in human sera. Gene Ther. 2016, 23, 592–596. [Google Scholar] [CrossRef]
- Atasheva, S.; Emerson, C.C.; Yao, J.; Young, C.; Stewart, P.L.; Shayakhmetov, D.M. Systemic cancer therapy with engineered adenovirus that evades innate immunity. Sci. Transl. Med. 2020, 12, eabc6659. [Google Scholar] [CrossRef]
- Dai, X.; Wu, L.; Sun, R.; Zhou, Z.H. Atomic Structures of Minor Proteins VI and VII in Human Adenovirus. J. Virol. 2017, 91, e00850-17. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Van Raaij, M.J.; Mitraki, A.; Lavigne, G.; Cusack, S. A triple beta-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 1999, 401, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Liu, H.; Wu, L.; Zhou, Z.H. Model of the trimeric fiber and its interactions with the pentameric penton base of human adenovirus by cryo-electron microscopy. J. Mol. Biol. 2011, 406, 764–774. [Google Scholar] [CrossRef]
- Muller, R.; Grawert, M.A.; Kern, T.; Madl, T.; Peschek, J.; Sattler, M.; Groll, M.; Buchner, J. High-resolution structures of the IgM Fc domains reveal principles of its hexamer formation. Proc. Natl. Acad. Sci. USA 2013, 110, 10183–10188. [Google Scholar] [CrossRef]
- Graille, M.; Stura, E.A.; Corper, A.L.; Sutton, B.J.; Taussig, M.J.; Charbonnier, J.B.; Silverman, G.J. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: Structural basis for recognition of B-cell receptors and superantigen activity. Proc. Natl. Acad. Sci. USA 2000, 97, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.A.; Sander, B.; Jensen, R.K.; Pedersen, J.S.; Golas, M.M.; Jensenius, J.C.; Hansen, A.G.; Thiel, S.; Andersen, G.R. Structure and activation of C1, the complex initiating the classical pathway of the complement cascade. Proc. Natl. Acad. Sci. USA 2017, 114, 986–991. [Google Scholar] [CrossRef]
- Almitairi, J.O.M.; Venkatraman Girija, U.; Furze, C.M.; Simpson-Gray, X.; Badakshi, F.; Marshall, J.E.; Schwaeble, W.J.; Mitchell, D.A.; Moody, P.C.E.; Wallis, R. Structure of the C1r-C1s interaction of the C1 complex of complement activation. Proc. Natl. Acad. Sci. USA 2018, 115, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Budayova-Spano, M.; Lacroix, M.; Thielens, N.M.; Arlaud, G.J.; Fontecilla-Camps, J.C.; Gaboriaud, C. The crystal structure of the zymogen catalytic domain of complement protease C1r reveals that a disruptive mechanical stress is required to trigger activation of the C1 complex. EMBO J. 2002, 21, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.J.; Wijeyewickrema, L.C.; Wilmann, P.G.; Gunzburg, M.J.; D’Andrea, L.; Irving, J.A.; Pang, S.S.; Duncan, R.C.; Wilce, J.A.; Whisstock, J.C.; et al. A molecular switch governs the interaction between the human complement protease C1s and its substrate, complement C4. J. Biol. Chem. 2013, 288, 15821–15829. [Google Scholar] [CrossRef] [PubMed]
- Croll, T.I.; Andersen, G.R. Re-evaluation of low-resolution crystal structures via interactive molecular-dynamics flexible fitting (iMDFF): A case study in complement C4. Acta Crystallogr. D Struct. Biol. 2016, 72 Pt 9, 1006–1016. [Google Scholar] [CrossRef]
- Forneris, F.; Wu, J.; Xue, X.; Ricklin, D.; Lin, Z.; Sfyroera, G.; Tzekou, A.; Volokhina, E.; Granneman, J.C.; Hauhart, R.; et al. Regulators of complement activity mediate inhibitory mechanisms through a common C3b-binding mode. EMBO J. 2016, 35, 1133–1149. [Google Scholar] [CrossRef]
- Laursen, N.S.; Andersen, K.R.; Braren, I.; Spillner, E.; Sottrup-Jensen, L.; Andersen, G.R. Substrate recognition by complement convertases revealed in the C5-cobra venom factor complex. EMBO J. 2011, 30, 606–616. [Google Scholar] [CrossRef]
- Sim, R.B.; Twose, T.M.; Paterson, D.S.; Sim, E. The covalent-binding reaction of complement component C3. Biochem. J. 1981, 193, 115–127. [Google Scholar] [CrossRef]
- Venkateswarlu, D.; Perera, L.; Darden, T.; Pedersen, L.G. Structure and dynamics of zymogen human blood coagulation factor X. Biophys. J. 2002, 82, 1190–1206. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Sercinoglu, O.; Ozbek, P. gRINN: A tool for calculation of residue interaction energies and protein energy network analysis of molecular dynamics simulations. Nucleic Acids Res. 2018, 46, W554–W562. [Google Scholar] [CrossRef] [PubMed]
- Saban, S.D.; Silvestry, M.; Nemerow, G.R.; Stewart, P.L. Visualization of alpha-helices in a 6-angstrom resolution cryoelectron microscopy structure of adenovirus allows refinement of capsid protein assignments. J. Virol. 2006, 80, 12049–12059. [Google Scholar] [CrossRef] [PubMed]
- Law, S.K.; Dodds, A.W. The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci. 1997, 6, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.; Kidmose, R.T.; Petersen, S.V.; Szilagyi, A.; Prohaszka, Z.; Andersen, G.R. Structural Basis for the Function of Complement Component C4 within the Classical and Lectin Pathways of Complement. J. Immunol. 2015, 194, 5488–5496. [Google Scholar] [CrossRef]
- Emerson, C.C.; Stewart, P.L. Structure-Based Modeling of Complement C4 Mediated Neutralization of Adenovirus. Viruses 2021, 13, 111. [Google Scholar] [CrossRef]
- Lindert, S.; Silvestry, M.; Mullen, T.M.; Nemerow, G.R.; Stewart, P.L. Cryo-electron microscopy structure of an adenovirus-integrin complex indicates conformational changes in both penton base and integrin. J. Virol. 2009, 83, 11491–11501. [Google Scholar] [CrossRef]
- Flatt, J.W.; Kim, R.; Smith, J.G.; Nemerow, G.R.; Stewart, P.L. An intrinsically disordered region of the adenovirus capsid is implicated in neutralization by human alpha defensin 5. PLoS ONE 2013, 8, e61571. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C3b Pose | Penton Base Chain P1 | Penton Base Chain P2 | Penton Base Chain P3 | Penton Base Chain P4 | Penton Base Chain P5 | Fiber Chain F1 | Fiber Chain F2 | Fiber Chain F3 |
|---|---|---|---|---|---|---|---|---|
| 1 | 0 | −305 | 0 | 0 | 0 | 0 | −198 | 0 |
| 2 | −58 | −52 | 0 | 0 | 0 | −10 | 0 | −173 |
| 3 | 0 | 0 | 0 | −75 | 0 | 9 | −123 | −47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, N.; Shayakhmetov, D.M.; Stewart, P.L. Structural Model for Factor X Inhibition of IgM and Complement-Mediated Neutralization of Adenovirus. Viruses 2023, 15, 1343. https://doi.org/10.3390/v15061343
Wagner N, Shayakhmetov DM, Stewart PL. Structural Model for Factor X Inhibition of IgM and Complement-Mediated Neutralization of Adenovirus. Viruses. 2023; 15(6):1343. https://doi.org/10.3390/v15061343
Chicago/Turabian StyleWagner, Nicole, Dmitry M. Shayakhmetov, and Phoebe L. Stewart. 2023. "Structural Model for Factor X Inhibition of IgM and Complement-Mediated Neutralization of Adenovirus" Viruses 15, no. 6: 1343. https://doi.org/10.3390/v15061343
APA StyleWagner, N., Shayakhmetov, D. M., & Stewart, P. L. (2023). Structural Model for Factor X Inhibition of IgM and Complement-Mediated Neutralization of Adenovirus. Viruses, 15(6), 1343. https://doi.org/10.3390/v15061343