
Effect of Viral Strain and Host Age on Clinical Disease and Viral Replication in Immunocompetent Mouse Models of Chikungunya Encephalomyelitis

Abstract
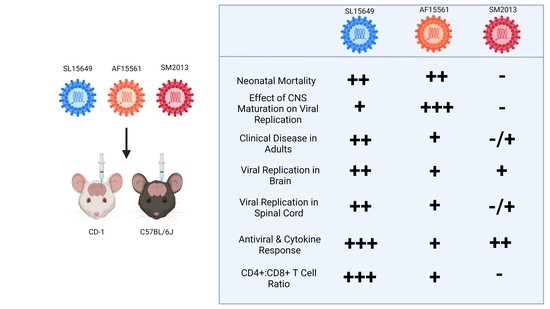
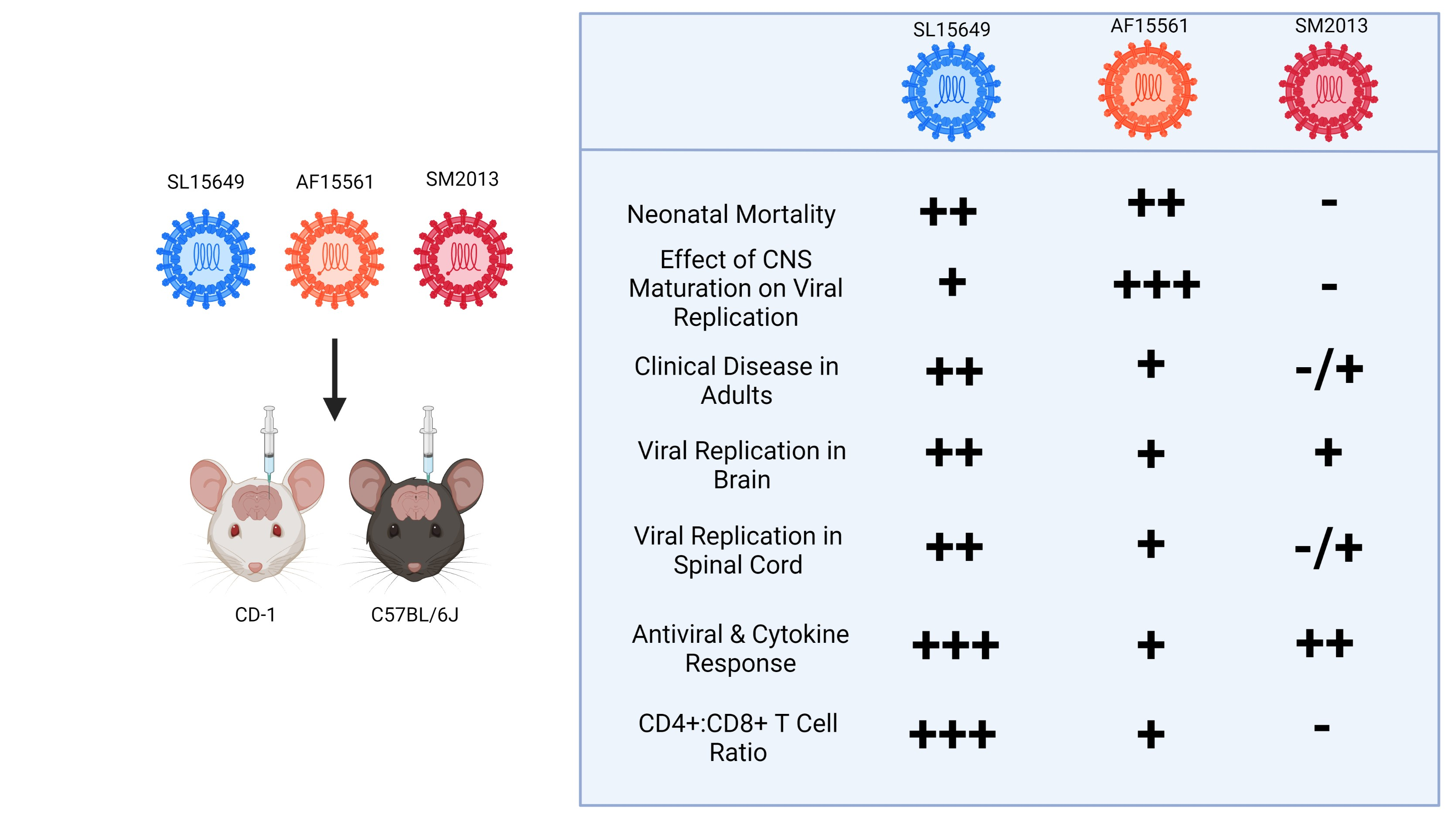{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Virus Stocks
2.3. Mouse Infections
2.4. Tissue Collection
2.5. Quantification of Infectious Virus
2.6. RNA Isolation and Quantitative PCR
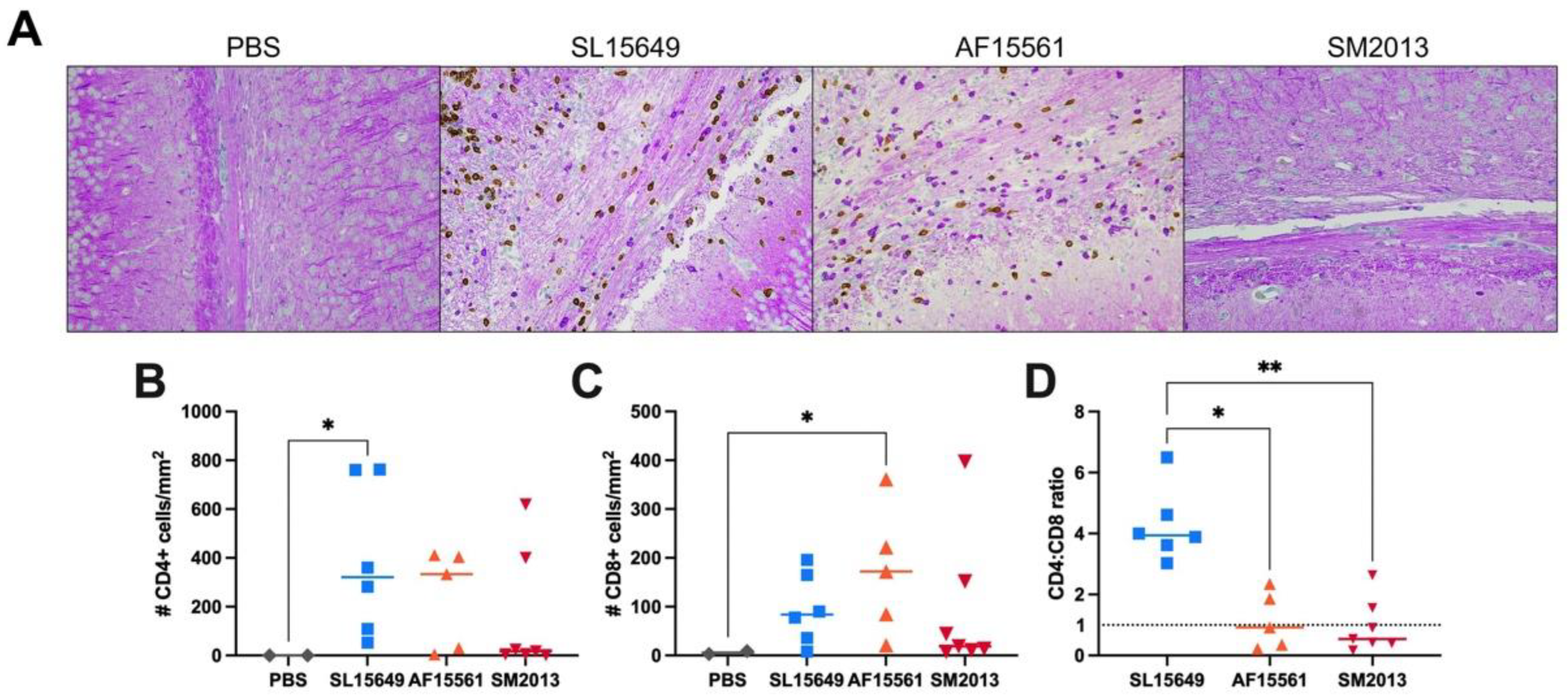
2.7. Histopathology
2.8. Statistics
3. Results
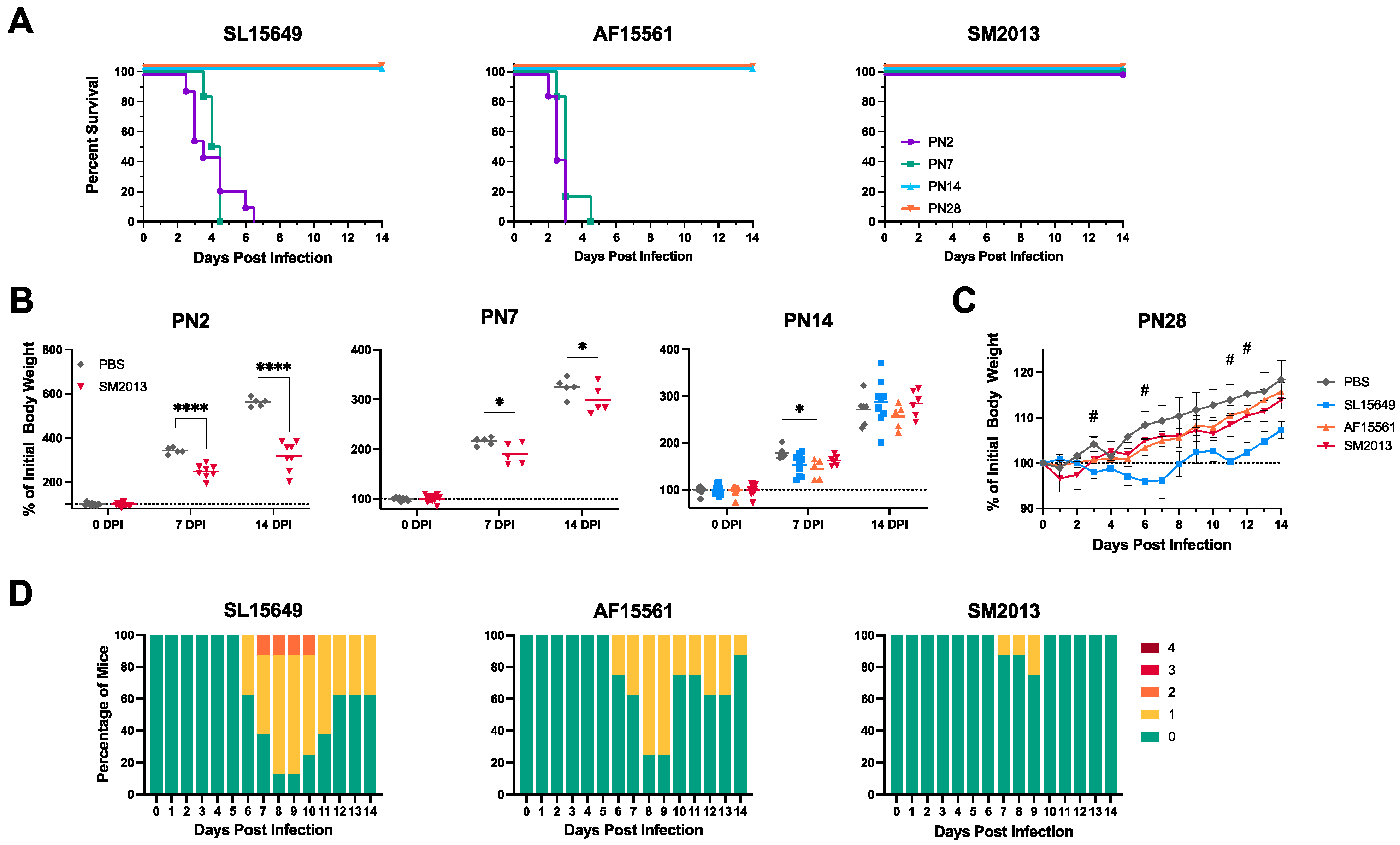
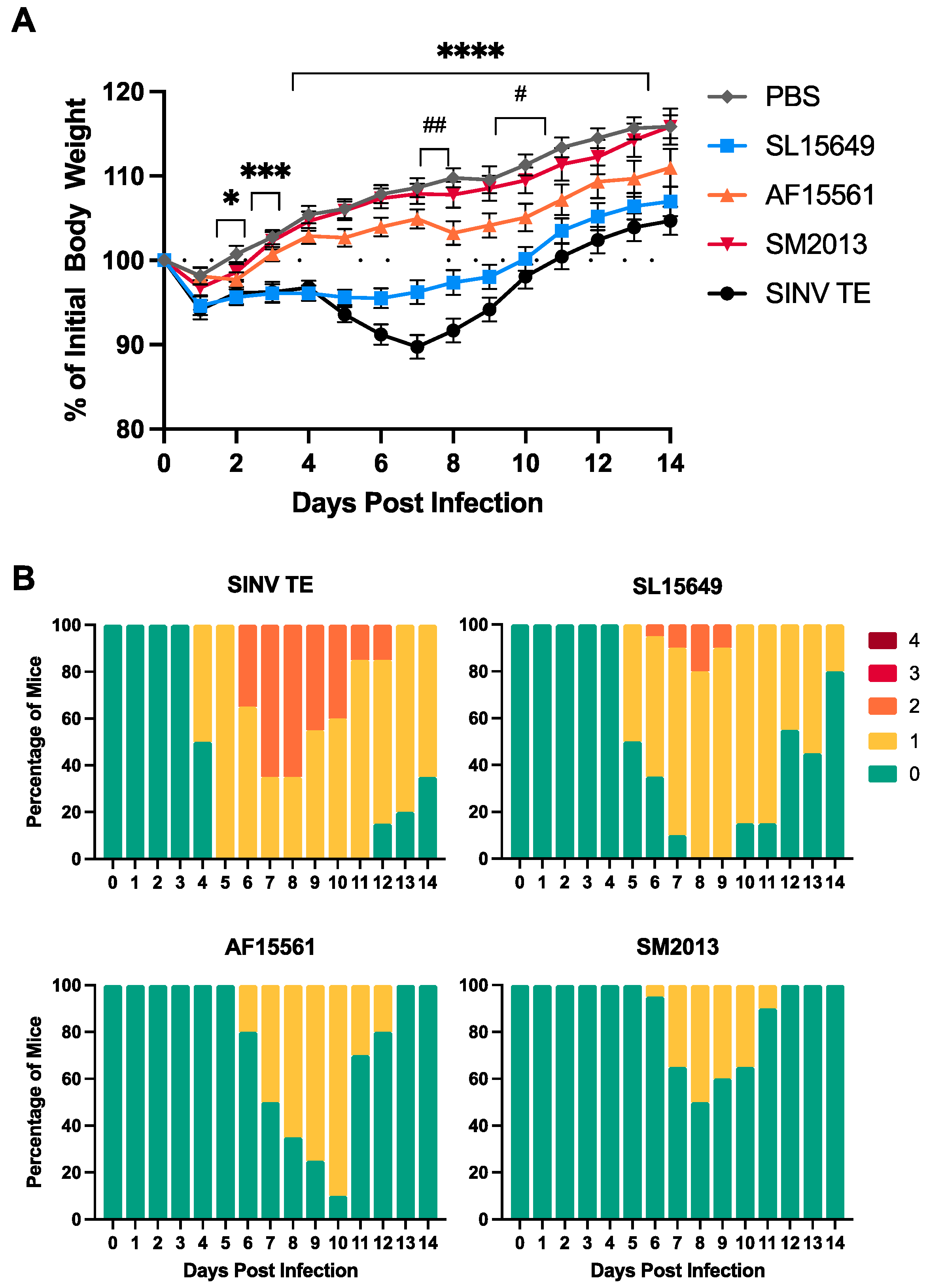
3.1. Outbred CD-1 Mice Demonstrate Age- and Viral Strain-Dependent Clinical Outcomes Following Intracranial CHIKV Infection
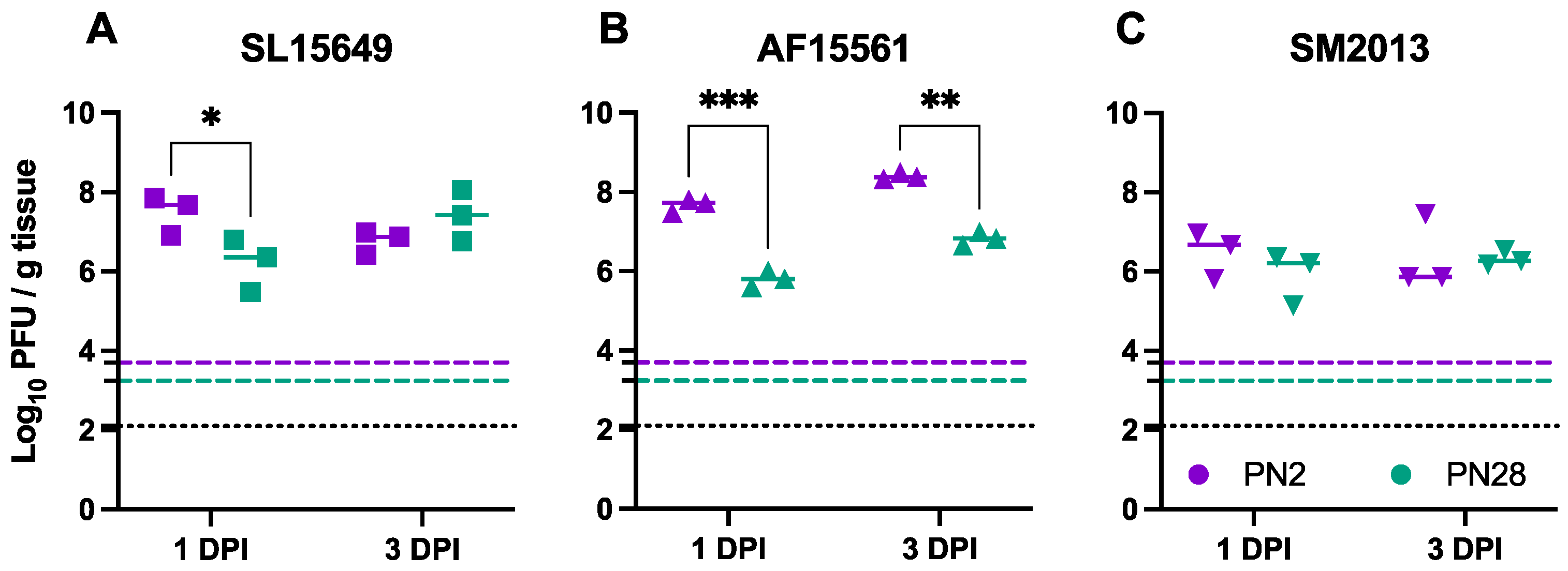
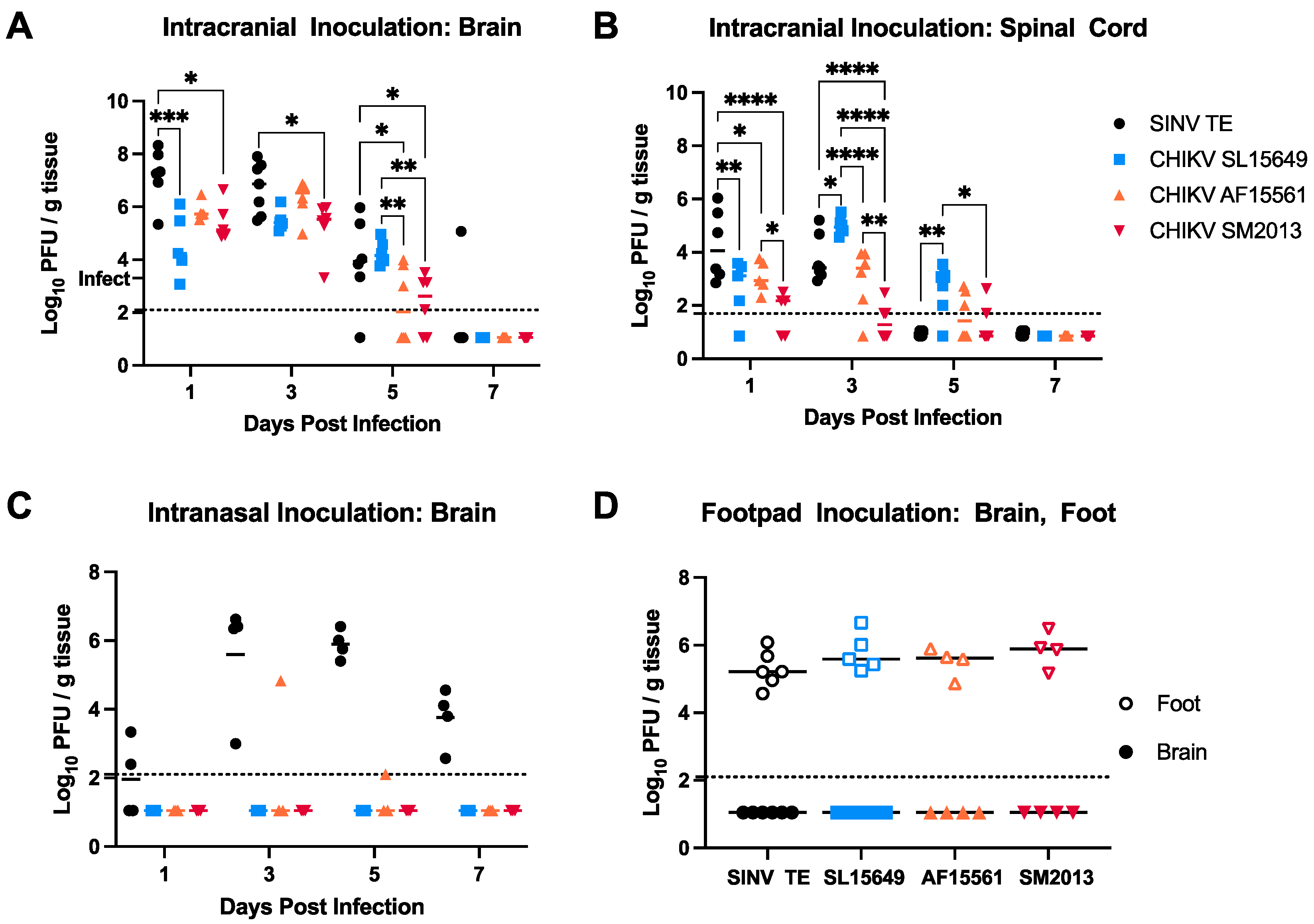
3.2. Viral Replication in the Brain Differs Based on Age of Infection and CHIKV Strain in CD-1 Mice
3.3. Four-To-Six-Week-Old C57BL/6J Mice Are Susceptible to CHIKV-Induced Clinical and Neurological Disease
3.4. Viral Titers in Brain and Spinal Cord Vary by CHIKV Strain and Inoculation Route in C57BL/6 Mice
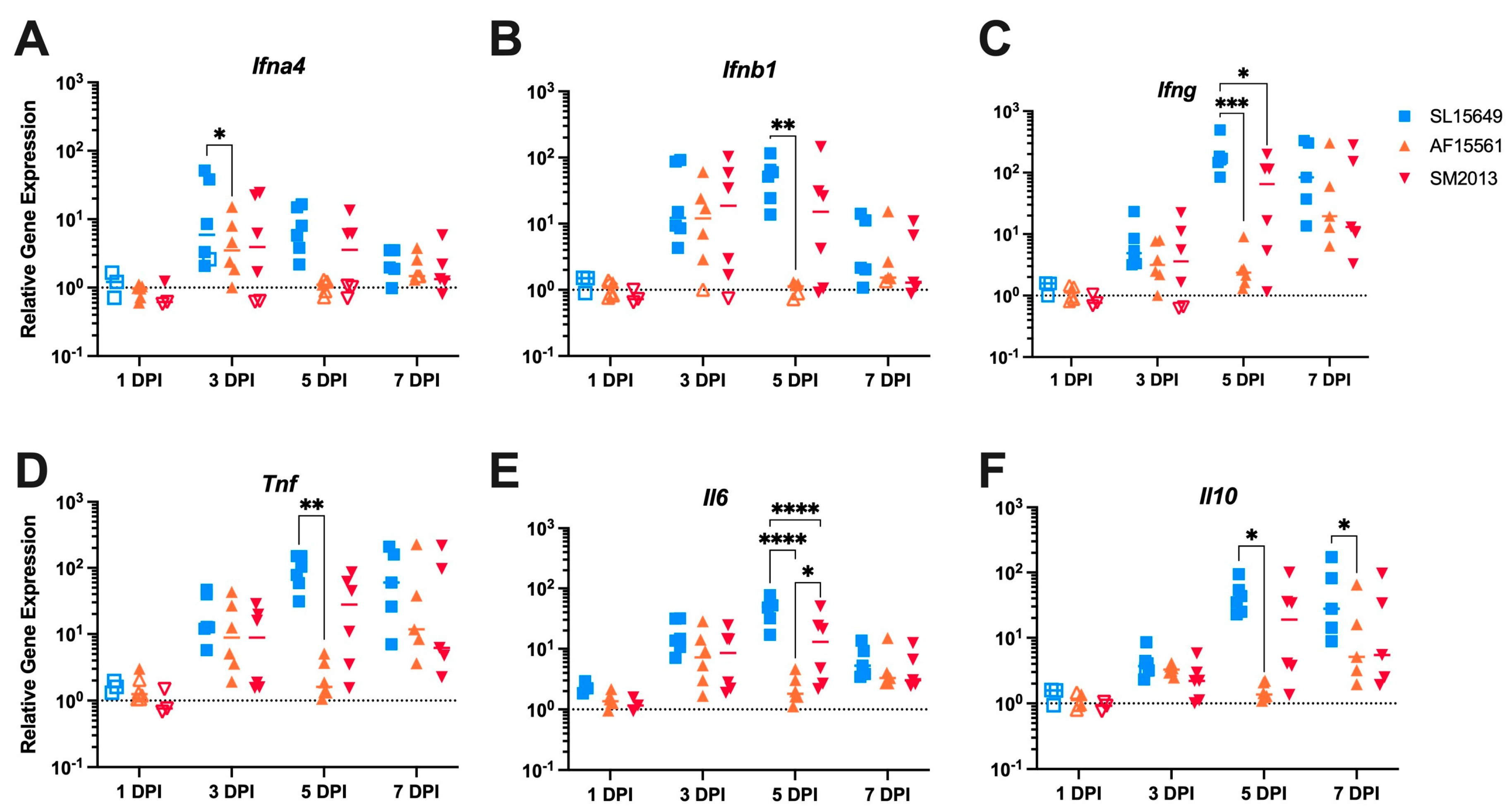
3.5. The Immune Response in the Brain to IC CHIKV Infection Is CHIKV Strain Dependent
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silva, L.A.; Dermody, T.S. Chikungunya Virus: Epidemiology, Replication, Disease Mechanisms, and Prospective Intervention Strategies. J. Clin. Investig. 2017, 127, 737–749. [Google Scholar] [CrossRef]
- Robinson, M.C. An Epidemic of Virus Disease in Southern Province, Tanganyika Territory, in 1952–1953. Trans. R. Soc. Trop. Med. Hyg. 1955, 49, 28–32. [Google Scholar] [CrossRef]
- Chandak, N.H.; Kashyap, R.S.; Kabra, D.; Karandikar, P.; Saha, S.S.; Morey, S.H.; Purohit, H.J.; Taori, G.M.; Daginawala, H.F. Neurological Complications of Chikungunya Virus Infection. Neurol. India 2009, 57, 177. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.A.R.; Ferreira, L.F.; Neto, R.D.J.P. Acute Disseminated Encephalomyelitis After Chikungunya Infection. JAMA Neurol. 2019, 76, 619. [Google Scholar] [CrossRef]
- Mehta, R.; Gerardin, P.; De Brito, C.A.A.; Soares, C.N.; Ferreira, M.L.B.; Solomon, T. The Neurological Complications of Chikungunya Virus: A Systematic Review. Rev. Med. Virol. 2018, 28, e1978. [Google Scholar] [CrossRef]
- Ramos, R.; Viana, R.; Brainer-Lima, A.; FloreÂncio, T.; Carvalho, M.D.; van der Linden, V.; Amorim, A.; Rocha, M.A.; Medeiros, F. Perinatal Chikungunya Virus–Associated Encephalitis Leading to Postnatal-Onset Microcephaly and Optic Atrophy. Pediatr. Infect. Dis. J. 2018, 37, 94–95. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Rehani, V.; Kumar, P.; Sasmal, G.; Goyal, P. Protean Neurological Manifestations in Chikungunya. J. Assoc. Physicians India 2018, 66, 79–80. [Google Scholar] [PubMed]
- Maria, A.; Vallamkonda, N.; Shukla, A.; Bhatt, A.; Sachdev, N. Encephalitic Presentation of Neonatal Chikungunya: A Case Series. Indian Pediatr. 2018, 55, 671–674. [Google Scholar] [CrossRef]
- Oehler, E.; Fournier, E.; Leparc-Goffart, I.; Larre, P.; Cubizolle, S.; Sookhareea, C.; Lastère, S.; Ghawche, F. Increase in Cases of Guillain-Barré Syndrome during a Chikungunya Outbreak, French Polynesia, 2014 to 2015. Eurosurveillance 2015, 20, 30079. [Google Scholar] [CrossRef]
- Robin, S.; Ramful, D.; Seach, F.L.; Jaffar-Bandjee, M.-C.; Rigou, G.; Alessandri, J.-L. Neurologic Manifestations of Pediatric Chikungunya Infection. J. Child Neurol. 2008, 23, 1028–1035. [Google Scholar] [CrossRef]
- Anand, K.S.; Agrawal, A.K.; Garg, J.; Dhamija, R.K.; Mahajan, R.K. Spectrum of Neurological Complications in Chikungunya Fever: Experience at a Tertiary Care Centre and Review of Literature. Trop. Dr. 2019, 49, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Cerny, T.; Schwarz, M.; Schwarz, U.; Lemant, J.; Gérardin, P.; Keller, E. The Range of Neurological Complications in Chikungunya Fever. Neurocrit. Care 2017, 27, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E. Role of the Immune Response in Age-Dependent Resistance of Mice to Encephalitis Due to Sindbis Virus. J. Infect. Dis. 1976, 133, 456–464. [Google Scholar] [CrossRef]
- Tucker, P.C.; Griffin, D.E. Mechanism of Altered Sindbis Virus Neurovirulence Associated with a Single-Amino-Acid Change in the E2 Glycoprotein. J. Virol. 1991, 65, 1551–1557. [Google Scholar] [CrossRef]
- Griffin, D.E. Alphavirus Encephalomyelitis: Mechanisms and Approaches to Prevention of Neuronal Damage. Neurotherapeutics 2016, 13, 455–460. [Google Scholar] [CrossRef]
- Wang, E.; Volkova, E.; Adams, A.P.; Forrester, N.; Xiao, S.-Y.; Frolov, I.; Weaver, S.C. Chimeric Alphavirus Vaccine Candidates for Chikungunya. Vaccine 2008, 26, 5030–5039. [Google Scholar] [CrossRef] [PubMed]
- Jain, J.; Narayanan, V.; Chaturvedi, S.; Pai, S.; Sunil, S. In Vivo Evaluation of Withania Somnifera–Based Indian Traditional Formulation (Amukkara Choornam), Against Chikungunya Virus–Induced Morbidity and Arthralgia. J. Evid. Based Integr. Med. 2018, 23, 2156587218757661. [Google Scholar] [CrossRef]
- Jain, J.; Narayanan, V.; Kumar, A.; Shrinet, J.; Srivastava, P.; Chaturvedi, S.; Sunil, S. Establishment and Comparison of Pathogenicity and Related Neurotropism in Two Age Groups of Immune Competent Mice, C57BL/6J Using an Indian Isolate of Chikungunya Virus (CHIKV). Viruses 2019, 11, 578. [Google Scholar] [CrossRef]
- Suckling, A.J.; Jagelman, S.; Webb, H.E. A Comparison of Brain Lysosomal Enzyme Activities in Four Experimental Togavirus Encephalitides. J. Neurol. Sci. 1978, 35, 355–364. [Google Scholar] [CrossRef]
- Teo, T.-H.; Lum, F.-M.; Lee, W.W.L.; Ng, L.F.P. Mouse Models for Chikungunya Virus: Deciphering Immune Mechanisms Responsible for Disease and Pathology. Immunol. Res. 2012, 53, 136–147. [Google Scholar] [CrossRef]
- Priya, R.; Patro, I.K.; Parida, M.M. TLR3 Mediated Innate Immune Response in Mice Brain Following Infection with Chikungunya Virus. Virus Res. 2014, 189, 194–205. [Google Scholar] [CrossRef]
- Ziegler, S.A.; Lu, L.; Xiao, S.-Y.; Da Rosa, A.P.A.T.; Tesh, R.B. An Animal Model for Studying the Pathogenesis of Chikungunya Virus Infection. Am. J. Trop. Med. Hyg. 2008, 79, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Chiam, C.W.; Chan, Y.F.; Ong, K.C.; Wong, K.T.; Sam, I.-C. Neurovirulence Comparison of Chikungunya Virus Isolates of the Asian and East/Central/South African Genotypes from Malaysia. J. Gen. Virol. 2015, 96, 3243–3254. [Google Scholar] [CrossRef]
- Couderc, T.; Chrétien, F.; Schilte, C.; Disson, O.; Brigitte, M.; Guivel-Benhassine, F.; Touret, Y.; Barau, G.; Cayet, N.; Schuffenecker, I.; et al. A Mouse Model for Chikungunya: Young Age and Inefficient Type-I Interferon Signaling Are Risk Factors for Severe Disease. PLoS Pathog. 2008, 4, e29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-L.; Dong, H.-L.; Zhang, Y.-N.; Xu, L.-L.; Deng, C.-L.; Li, X.-F.; Li, X.-D.; Ye, H.-Q.; Yuan, Z.-M.; Qin, C.-F.; et al. Visualization of Chikungunya Virus Infection in Vitro and in Vivo. Emerg. Microbes Infect. 2019, 8, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Precious, S.W.; Webb, H.E.; Bowen, E.T.W. Isolation and Persistence of Chikungunya Virus in Cultures of Mouse Brain Cells. J. Gen. Virol. 1974, 23, 271–279. [Google Scholar] [CrossRef]
- Chatterjee, S.N.; Sarkar, J.K. Electron Microscopic Studies of Suckling Mouse Brain Cells Infected with Chikungunya Virus. Indian J. Exp. Biol. 1965, 3, 227–234. [Google Scholar]
- Das, T.; Hoarau, J.J.; Bandjee, M.C.J.; Maquart, M.; Gasque, P. Multifaceted Innate Immune Responses Engaged by Astrocytes, Microglia and Resident Dendritic Cells against Chikungunya Neuroinfection. J. Gen. Virol. 2015, 96, 294–310. [Google Scholar] [CrossRef] [PubMed]
- Fraisier, C.; Koraka, P.; Belghazi, M.; Bakli, M.; Granjeaud, S.; Pophillat, M.; Lim, S.M.; Osterhaus, A.; Martina, B.; Camoin, L.; et al. Kinetic Analysis of Mouse Brain Proteome Alterations Following Chikungunya Virus Infection before and after Appearance of Clinical Symptoms. PLoS ONE 2014, 9, e91397. [Google Scholar] [CrossRef]
- Lim, S.M.; van den Ham, H.-J.; Oduber, M.; Martina, E.; Zaaraoui-Boutahar, F.; Roose, J.M.; van IJcken, W.F.J.; Osterhaus, A.D.M.E.; Andeweg, A.C.; Koraka, P.; et al. Transcriptomic Analyses Reveal Differential Gene Expression of Immune and Cell Death Pathways in the Brains of Mice Infected with West Nile Virus and Chikungunya Virus. Front. Microbiol. 2017, 8, 1556. [Google Scholar] [CrossRef]
- Dhanwani, R.; Khan, M.; Alam, S.I.; Rao, P.V.L.; Parida, M. Differential Proteome Analysis of Chikungunya Virus-infected New-born Mice Tissues Reveal Implication of Stress, Inflammatory and Apoptotic Pathways in Disease Pathogenesis. Proteomics 2011, 11, 1936–1951. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.R.; Abraham, R.; Sundaram, S.; Sreekumar, E. Interferon Regulated Gene (IRG) Expression-Signature in a Mouse Model of Chikungunya Virus Neurovirulence. J. Neurovirol. 2017, 23, 886–902. [Google Scholar] [CrossRef]
- Tun, M.M.N.; Muthugala, R.; Kyaw, A.K.; Shimada, S.; Morita, K.; Hayasaka, D. Pathogenetic Potential Relating to Metabolic Activity in a Mouse Model of Infection with the Chikungunya Virus East/Central/South African Genotype. Viruses 2020, 12, 169. [Google Scholar] [CrossRef]
- Levitt, N.H.; Ramsburg, H.H.; Hasty, S.E.; Repik, P.M.; Cole, F.E.; Lupton, H.W. Development of an Attenuated Strain of Chikungunya Virus for Use in Vaccine Production. Vaccine 1986, 4, 157–162. [Google Scholar] [CrossRef]
- Powers, A.M.; Logue, C.H. Changing Patterns of Chikungunya Virus: Re-Emergence of a Zoonotic Arbovirus. J. Gen. Virol. 2007, 88, 2363–2377. [Google Scholar] [CrossRef] [PubMed]
- Diallo, M.; Thonnon, J.; Traore-Lamizana, M.; Fontenille, D. Vectors of Chikungunya Virus in Senegal: Current Data and Transmission Cycles. Am. J. Trop. Med. Hyg. 1999, 60, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Pistone, T.; Ezzedine, K.; Boisvert, M.; Receveur, M.; Schuffenecker, I.; Zeller, H.; Lafon, M.; Fleury, H.; Malvy, D. Cluster of Chikungunya Virus Infection in Travelers Returning from Senegal, 2006. J. Travel Med. 2009, 16, 286–288. [Google Scholar] [CrossRef]
- Julander, J.G.; Dagley, A.; Gebre, M.; Komeno, T.; Nakajima, N.; Smee, D.F.; Furuta, Y. Strain-Dependent Disease and Response to Favipiravir Treatment in Mice Infected with Chikungunya Virus. Antivir. Res. 2020, 182, 104904. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-I.; Reisen, W.K.; Brault, A.C.; Pesavento, P.; Clark, D.C.; Luciw, P.A.; Lerche, N.W. Comparative Pathogenesis of Epidemic and Enzootic Chikungunya Viruses in a Pregnant Rhesus Macaque Model. Am. J. Trop. Med. Hyg. 2010, 83, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Burt, F.J.; Chen, W.; Miner, J.J.; Lenschow, D.J.; Merits, A.; Schnettler, E.; Kohl, A.; Rudd, P.A.; Taylor, A.; Herrero, L.J.; et al. Chikungunya Virus: An Update on the Biology and Pathogenesis of This Emerging Pathogen. Lancet Infect. Dis. 2017, 17, e107–e117. [Google Scholar] [CrossRef]
- Weaver, S.C.; Forrester, N.L. Chikungunya: Evolutionary History and Recent Epidemic Spread. Antivir. Res. 2015, 120, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.; Rousset, D.; Huc, P.; Matheus, S.; Ledrans, M.; Rosine, J.; Cassadou, S.; Noël, H. Seroprevalence of Asian Lineage Chikungunya Virus Infection on Saint Martin Island, 7 Months after the 2013 Emergence. Am. J. Trop. Med. Hyg. 2016, 94, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Presti, A.L.; Lai, A.; Cella, E.; Zehender, G.; Ciccozzi, M. Chikungunya Virus, Epidemiology, Clinics and Phylogenesis: A Review. Asian Pac. J. Trop. Med. 2014, 7, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Langsjoen, R.M.; Haller, S.L.; Roy, C.J.; Vinet-Oliphant, H.; Bergren, N.A.; Erasmus, J.H.; Livengood, J.A.; Powell, T.D.; Weaver, S.C.; Rossi, S.L. Chikungunya Virus Strains Show Lineage-Specific Variations in Virulence and Cross-Protective Ability in Murine and Nonhuman Primate Models. Mbio 2018, 9, e02449-17. [Google Scholar] [CrossRef]
- Gérardin, P.; Freitas, A.R.R.; Sissoko, D.; Teixeira, M.G. Transmission Dynamics and Disease Severity in Children Infected with East Central South African (ECSA) or ECSA-Diverged Clades of Chikungunya Virus. Clin. Infect. Dis. 2018, 68, 171–172. [Google Scholar] [CrossRef]
- Baxter, V.K.; Troisi, E.M.; Pate, N.M.; Zhao, J.N.; Griffin, D.E. Death and Gastrointestinal Bleeding Complicate Encephalomyelitis in Mice with Delayed Appearance of CNS IgM after Intranasal Alphavirus Infection. J. Gen. Virol. 2018, 99, 309–320. [Google Scholar] [CrossRef]
- Johnson, R.T.; McFarland, H.F.; Levy, S.E. Age-Dependent Resistance to Viral Encephalitis: Studies of Infections Due to Sindbis Virus in Mice. J. Infect. Dis. 1972, 125, 257–262. [Google Scholar] [CrossRef]
- Oliver, K.R.; Scallan, M.F.; Dyson, H.; Fazakerley, J.K. Susceptibility to a Neurotropic Virus and Its Changing Distribution in the Developing Brain Is a Function of CNS Maturity. J. Neurovirol. 1997, 3, 38–48. [Google Scholar] [CrossRef]
- Morrison, T.E.; Oko, L.; Montgomery, S.A.; Whitmore, A.C.; Lotstein, A.R.; Gunn, B.M.; Elmore, S.A.; Heise, M.T. A Mouse Model of Chikungunya Virus–Induced Musculoskeletal Inflammatory Disease Evidence of Arthritis, Tenosynovitis, Myositis, and Persistence. Am. J. Pathol. 2011, 178, 32–40. [Google Scholar] [CrossRef]
- Madden, E.A.; Plante, K.S.; Morrison, C.R.; Kutchko, K.M.; Sanders, W.; Long, K.M.; Taft-Benz, S.; Cisneros, M.C.C.; White, A.M.; Sarkar, S.; et al. Using SHAPE-MaP to Model RNA Secondary Structure and Identify 3′UTR Variation in Chikungunya Virus. J. Virol. 2020, 94, e00701-20. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T. Effects of Litter Size on Behavioral Development in Mice. Reprod. Toxicol. 1998, 12, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Martín-Sánchez, A.; Valera-Marín, G.; Hernández-Martínez, A.; Lanuza, E.; Martínez-García, F.; Agustín-Pavón, C. Wired for Motherhood: Induction of Maternal Care but Not Maternal Aggression in Virgin Female CD1 Mice. Front. Behav. Neurosci. 2015, 9, 197. [Google Scholar] [CrossRef] [PubMed]
- Labrada, L.; Liang, X.H.; Zheng, W.; Johnston, C.; Levine, B. Age-Dependent Resistance to Lethal Alphavirus Encephalitis in Mice: Analysis of Gene Expression in the Central Nervous System and Identification of a Novel Interferon-Inducible Protective Gene, Mouse ISG12. J. Virol. 2002, 76, 11688–11703. [Google Scholar] [CrossRef]
- Griffin, D.E.; Levine, B.; Tyor, W.R.; Tucker, P.C.; Hardwick, J.M. Age-Dependent Susceptibility to Fatal Encephalitis: Alphavirus Infection of Neurons. Arch. Virol. Suppl. 1994, 9, 31–39. [Google Scholar] [CrossRef]
- Morrison, T.E.; Whitmore, A.C.; Shabman, R.S.; Lidbury, B.A.; Mahalingam, S.; Heise, M.T. Characterization of Ross River Virus Tropism and Virus-Induced Inflammation in a Mouse Model of Viral Arthritis and Myositis. J. Virol. 2006, 80, 737–749. [Google Scholar] [CrossRef]
- Mesquita, L.P.; Costa, R.C.; Zanatto, D.A.; Bruhn, F.R.P.; Mesquita, L.L.R.; Lara, M.C.C.S.H.; Villalobos, E.M.C.; Massoco, C.O.; Mori, C.M.C.; Mori, E.; et al. Equine Herpesvirus 1 Elicits a Strong Pro-Inflammatory Response in the Brain of Mice. J. Gen. Virol. 2021, 102, 001556. [Google Scholar] [CrossRef]
- Garcia-Tapia, D.; Hassett, D.E.; Mitchell, W.J.; Johnson, G.C.; Kleiboeker, S.B. West Nile Virus Encephalitis: Sequential Histopathological and Immunological Events in a Murine Model of Infection. J. Neurovirol. 2007, 13, 130–138. [Google Scholar] [CrossRef]
- Atasheva, S.; Wang, E.; Adams, A.P.; Plante, K.S.; Ni, S.; Taylor, K.; Miller, M.E.; Frolov, I.; Weaver, S.C. Chimeric Alphavirus Vaccine Candidates Protect Mice from Intranasal Challenge with Western Equine Encephalitis Virus. Vaccine 2009, 27, 4309–4319. [Google Scholar] [CrossRef]
- Ronca, S.E.; Smith, J.; Koma, T.; Miller, M.M.; Yun, N.; Dineley, K.T.; Paessler, S. Mouse Model of Neurological Complications Resulting from Encephalitic Alphavirus Infection. Front. Microbiol. 2017, 8, 188. [Google Scholar] [CrossRef]
- Brooke, C.B.; Schäfer, A.; Matsushima, G.K.; White, L.J.; Johnston, R.E. Early Activation of the Host Complement System Is Required to Restrict Central Nervous System Invasion and Limit Neuropathology during Venezuelan Equine Encephalitis Virus Infection. J. Gen. Virol. 2012, 93, 797–806. [Google Scholar] [CrossRef]
- Passoni, G.; Langevin, C.; Palha, N.; Mounce, B.C.; Briolat, V.; Affaticati, P.; Job, E.D.; Joly, J.-S.; Vignuzzi, M.; Saleh, M.-C.; et al. Imaging of Viral Neuroinvasion in the Zebrafish Reveals That Sindbis and Chikungunya Viruses Favour Different Entry Routes. Dis. Model. Mech. 2017, 10, 847–857. [Google Scholar] [CrossRef]
- Cook, S.H.; Griffin, D.E. Luciferase Imaging of a Neurotropic Viral Infection in Intact Animals. J. Virol. 2003, 77, 5333–5338. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Griffin, D.E. Extensive Immune-Mediated Hippocampal Damage in Mice Surviving Infection with Neuroadapted Sindbis Virus. Virology 2003, 311, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Teo, T.-H.; Lum, F.-M.; Claser, C.; Lulla, V.; Lulla, A.; Merits, A.; Rénia, L.; Ng, L.F.P. A Pathogenic Role for CD4+ T Cells during Chikungunya Virus Infection in Mice. J. Immunol. 2013, 190, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Carissimo, G.; Teo, T.-H.; Chan, Y.-H.; Lee, C.Y.-P.; Lee, B.; Torres-Ruesta, A.; Tan, J.J.; Chua, T.-K.; Fong, S.-W.; Lum, F.-M.; et al. Viperin Controls Chikungunya Virus–Specific Pathogenic T Cell IFNγ Th1 Stimulation in Mice. Life Sci. Alliance 2019, 2, e201900298. [Google Scholar] [CrossRef]
- Fazakerley, J.K.; Webb, H.E. Semliki Forest Virus Induced, Immune Mediated Demyelination: The Effect of Irradiation. Br. J. Exp. Pathol. 1987, 68, 101–113. [Google Scholar]
- Baxter, V.K.; Glowinski, R.; Braxton, A.M.; Potter, M.C.; Slusher, B.S.; Griffin, D.E. Glutamine Antagonist-Mediated Immune Suppression Decreases Pathology but Delays Virus Clearance in Mice during Nonfatal Alphavirus Encephalomyelitis. Virology 2017, 508, 134–149. [Google Scholar] [CrossRef]
- Baxter, V.K.; Griffin, D.E. Interferon-Gamma Modulation of the Local T Cell Response to Alphavirus Encephalomyelitis. Viruses 2020, 12, 113. [Google Scholar] [CrossRef]
- Labadie, K.; Larcher, T.; Joubert, C.; Mannioui, A.; Delache, B.; Brochard, P.; Guigand, L.; Dubreil, L.; Lebon, P.; Verrier, B.; et al. Chikungunya Disease in Nonhuman Primates Involves Long-Term Viral Persistence in Macrophages. J. Clin. Investig. 2010, 120, 894–906. [Google Scholar] [CrossRef]
- Inglis, F.M.; Lee, K.M.; Chiu, K.B.; Purcell, O.M.; Didier, P.J.; Russell-Lodrigue, K.; Weaver, S.C.; Roy, C.J.; MacLean, A.G. Neuropathogenesis of Chikungunya Infection: Astrogliosis and Innate Immune Activation. J. Neurovirol. 2016, 22, 140–148. [Google Scholar] [CrossRef]
- Palha, N.; Guivel-Benhassine, F.; Briolat, V.; Lutfalla, G.; Sourisseau, M.; Ellett, F.; Wang, C.-H.; Lieschke, G.J.; Herbomel, P.; Schwartz, O.; et al. Real-Time Whole-Body Visualization of Chikungunya Virus Infection and Host Interferon Response in Zebrafish. PLoS Pathog. 2013, 9, e1003619. [Google Scholar] [CrossRef] [PubMed]
- Workman, A.D.; Charvet, C.J.; Clancy, B.; Darlington, R.B.; Finlay, B.L. Modeling Transformations of Neurodevelopmental Sequences across Mammalian Species. J. Neurosci. 2013, 33, 7368–7383. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.J.; Simpson, E.M.; Takahashi, J.S.; Lipp, H.-P.; Nakanishi, S.; Wehner, J.M.; Giese, K.P.; Tully, T.; Abel, T.; Chapman, P.F.; et al. Mutant Mice and Neuroscience: Recommendations Concerning Genetic Background. Neuron 1997, 19, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.R.; Fazakerley, J.K. Transneuronal Spread of Semliki Forest Virus in the Developing Mouse Olfactory System Is Determined by Neuronal Maturity. Neuroscience 1998, 82, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.L.W.; Vernon, P.S.; Griffin, D.E. Differentiation of Neurons Restricts Arbovirus Replication and Increases Expression of the Alpha Isoform of IRF-7. J. Virol. 2014, 89, 48–60. [Google Scholar] [CrossRef]
- Vernon, P.S.; Griffin, D.E. Characterization of an In Vitro Model of Alphavirus Infection of Immature and Mature Neurons. J. Virol. 2005, 79, 3438–3447. [Google Scholar] [CrossRef]
- Baxter, V.K.; Heise, M.T. Immunopathogenesis of Alphaviruses. Adv. Virus Res. 2020, 107, 315–382. [Google Scholar] [CrossRef]
- Jackson, A.C.; Moench, T.R.; Griffin, D.E.; Johnson, R.T. The Pathogenesis of Spinal Cord Involvement in the Encephalomyelitis of Mice Caused by Neuroadapted Sindbis Virus Infection. Lab. Investig. 1987, 56, 418–423. [Google Scholar]
- Kulcsar, K.A.; Baxter, V.K.; Greene, I.P.; Griffin, D.E. Interleukin 10 Modulation of Pathogenic Th17 Cells during Fatal Alphavirus Encephalomyelitis. Proc. Natl. Acad. Sci. USA 2014, 111, 16053–16058. [Google Scholar] [CrossRef]
- Carmen, J.; Rothstein, J.D.; Kerr, D.A. Tumor Necrosis Factor-α Modulates Glutamate Transport in the CNS and Is a Critical Determinant of Outcome from Viral Encephalomyelitis. Brain Res. 2009, 1263, 143–154. [Google Scholar] [CrossRef]
- Sarid, R.; Ben-Moshe, T.; Kazimirsky, G.; Weisberg, S.; Appel, E.; Kobiler, D.; Lustig, S.; Brodie, C. VFLIP Protects PC-12 Cells from Apoptosis Induced by Sindbis Virus: Implications for the Role of TNF-α. Cell Death Differ. 2001, 8, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Baxter, V.K.; Griffin, D.E. Interferon Gamma Modulation of Disease Manifestation and the Local Antibody Response to Alphavirus Encephalomyelitis. J. Gen. Virol. 2016, 97, 2908–2925. [Google Scholar] [CrossRef] [PubMed]
- Rowell, J.F.; Griffin, D.E. Contribution of T Cells to Mortality in Neurovirulent Sindbis Virus Encephalomyelitis. J. Neuroimmunol. 2002, 127, 106–114. [Google Scholar] [CrossRef]
- Mapalagamage, M.; Weiskopf, D.; Sette, A.; Silva, A.D.D. Current Understanding of the Role of T Cells in Chikungunya, Dengue and Zika Infections. Viruses 2022, 14, 242. [Google Scholar] [CrossRef] [PubMed]
- Lustig, S.; Jackson, A.C.; Hahn, C.S.; Griffin, D.E.; Strauss, E.G.; Strauss, J.H. Molecular Basis of Sindbis Virus Neurovirulence in Mice. J. Virol. 1988, 62, 2329–2336. [Google Scholar] [CrossRef]
- Ludwig, G.V.; Turell, M.J.; Vogel, P.; Kondig, J.P.; Kell, W.K.; Smith, J.F.; Pratt, W.D. Comparative Neurovirulence of Attenuated and Non-Attenuated Strains of Venezuelan Equine Encephalitis Virus in Mice. Am. J. Trop. Med. Hyg. 2001, 64, 49–55. [Google Scholar] [CrossRef]
- Lustig, S.; Halevy, M.; Ben-Nathan, D.; Akov, Y. A Novel Variant of Sindbis Virus Is Both Neurovirulent and Neuroinvasive in Adult Mice. Arch. Virol. 1992, 122, 237–248. [Google Scholar] [CrossRef]
- Boone, L.R.; Brown, A. Variants of the HR Strain of Sindbis Virus Lethal for Mice. J. Gen. Virol. 1976, 31, 261–263. [Google Scholar] [CrossRef]
- Griffin, D.E.; Johnson, R.T. Role of the Immune Response in Recovery from Sindbis Virus Encephalitis in Mice. J. Immunol. 1977, 118, 1070–1075. [Google Scholar] [CrossRef]
- Ferguson, M.C.; Saul, S.; Fragkoudis, R.; Weisheit, S.; Cox, J.; Patabendige, A.; Sherwood, K.; Watson, M.; Merits, A.; Fazakerley, J.K. Ability of the Encephalitic Arbovirus Semliki Forest Virus to Cross the Blood-Brain Barrier Is Determined by the Charge of the E2 Glycoprotein. J. Virol. 2015, 89, 7536–7549. [Google Scholar] [CrossRef]
- Dubuisson, J.; Lustig, S.; Ruggli, N.; Akov, Y.; Rice, C.M. Genetic Determinants of Sindbis Virus Neuroinvasiveness. J. Virol. 1997, 71, 2636–2646. [Google Scholar] [CrossRef] [PubMed]
- Lustig, S.; Danenberg, H.D.; Kafri, Y.; Kobiler, D.; Ben-Nathan, D. Viral Neuroinvasion and Encephalitis Induced by Lipopolysaccharide and Its Mediators. J. Exp. Med. 1992, 176, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Grauer, E.; Nathan, D.B.; Lustig, S.; Kobiler, D.; Kapon, J.; Danenberg, H.D. Viral Neuroinvasion as a Marker for BBB Integrity Following Exposure to Cholinesterase Inhibitors. Life Sci. 2001, 68, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Brooke, C.B.; Whitmore, A.C.; Johnston, R.E. The Role of the Blood-Brain Barrier during Venezuelan Equine Encephalitis Virus Infection. J. Virol. 2011, 85, 10682–10690. [Google Scholar] [CrossRef] [PubMed]
- Cain, M.D.; Salimi, H.; Gong, Y.; Yang, L.; Hamilton, S.L.; Heffernan, J.R.; Hou, J.; Miller, M.J.; Klein, R.S. Virus Entry and Replication in the Brain Precedes Blood-Brain Barrier Disruption during Intranasal Alphavirus Infection. J. Neuroimmunol. 2017, 308, 118–130. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, E.J.; Knight, A.C.; Heise, M.T.; Baxter, V.K. Effect of Viral Strain and Host Age on Clinical Disease and Viral Replication in Immunocompetent Mouse Models of Chikungunya Encephalomyelitis. Viruses 2023, 15, 1057. https://doi.org/10.3390/v15051057
Anderson EJ, Knight AC, Heise MT, Baxter VK. Effect of Viral Strain and Host Age on Clinical Disease and Viral Replication in Immunocompetent Mouse Models of Chikungunya Encephalomyelitis. Viruses. 2023; 15(5):1057. https://doi.org/10.3390/v15051057
Chicago/Turabian StyleAnderson, Elizabeth J., Audrey C. Knight, Mark T. Heise, and Victoria K. Baxter. 2023. "Effect of Viral Strain and Host Age on Clinical Disease and Viral Replication in Immunocompetent Mouse Models of Chikungunya Encephalomyelitis" Viruses 15, no. 5: 1057. https://doi.org/10.3390/v15051057
APA StyleAnderson, E. J., Knight, A. C., Heise, M. T., & Baxter, V. K. (2023). Effect of Viral Strain and Host Age on Clinical Disease and Viral Replication in Immunocompetent Mouse Models of Chikungunya Encephalomyelitis. Viruses, 15(5), 1057. https://doi.org/10.3390/v15051057