
A Perspective on Current Flavivirus Vaccine Development: A Brief Review

Abstract
1. Introduction
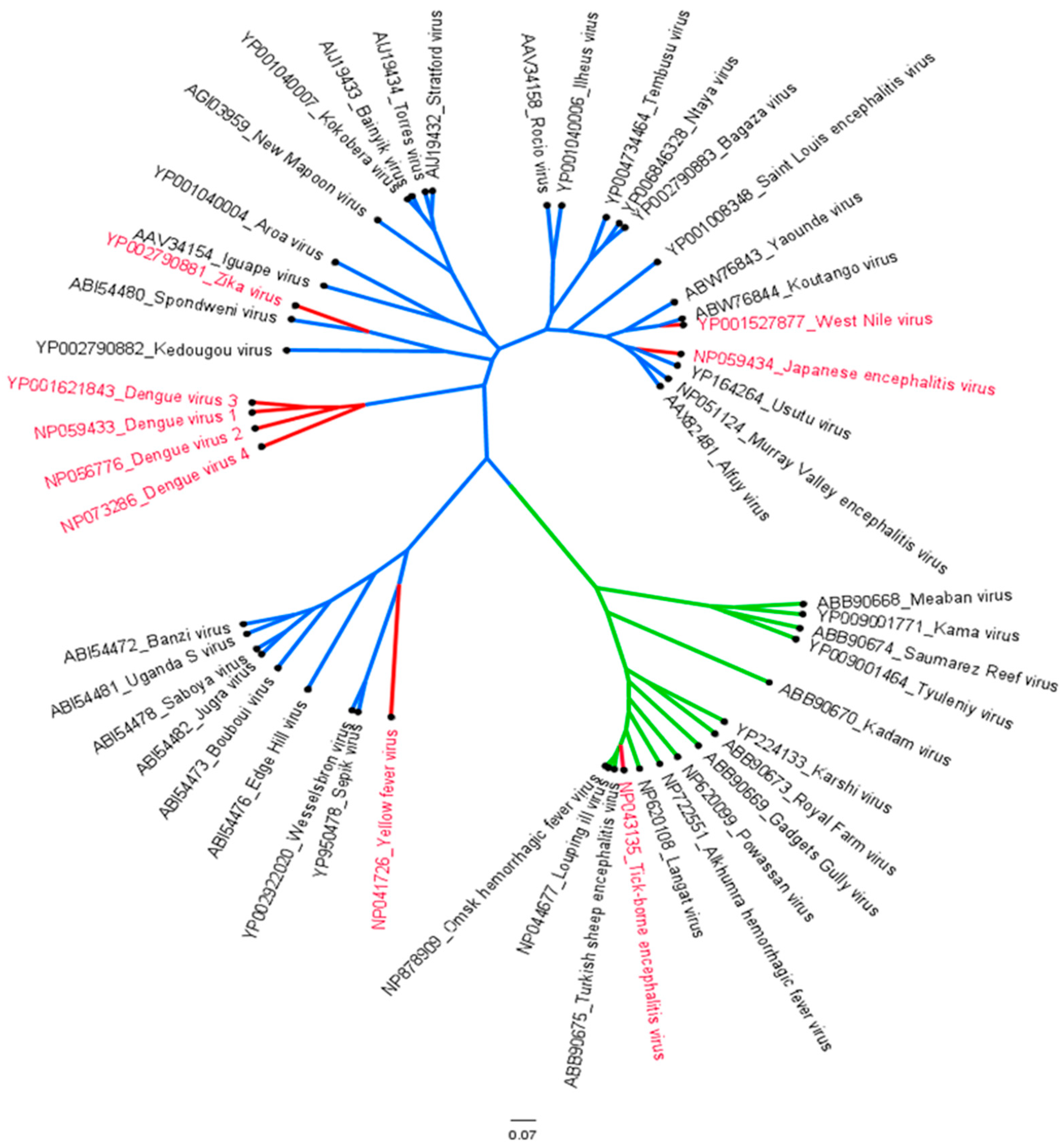
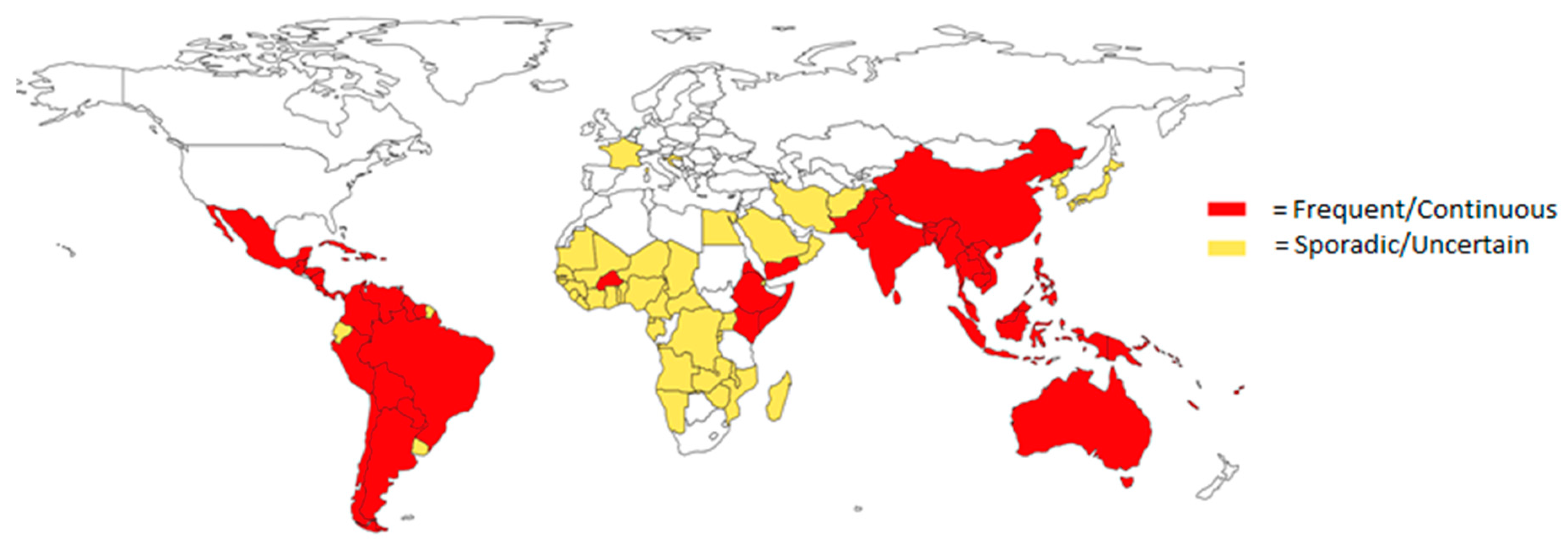
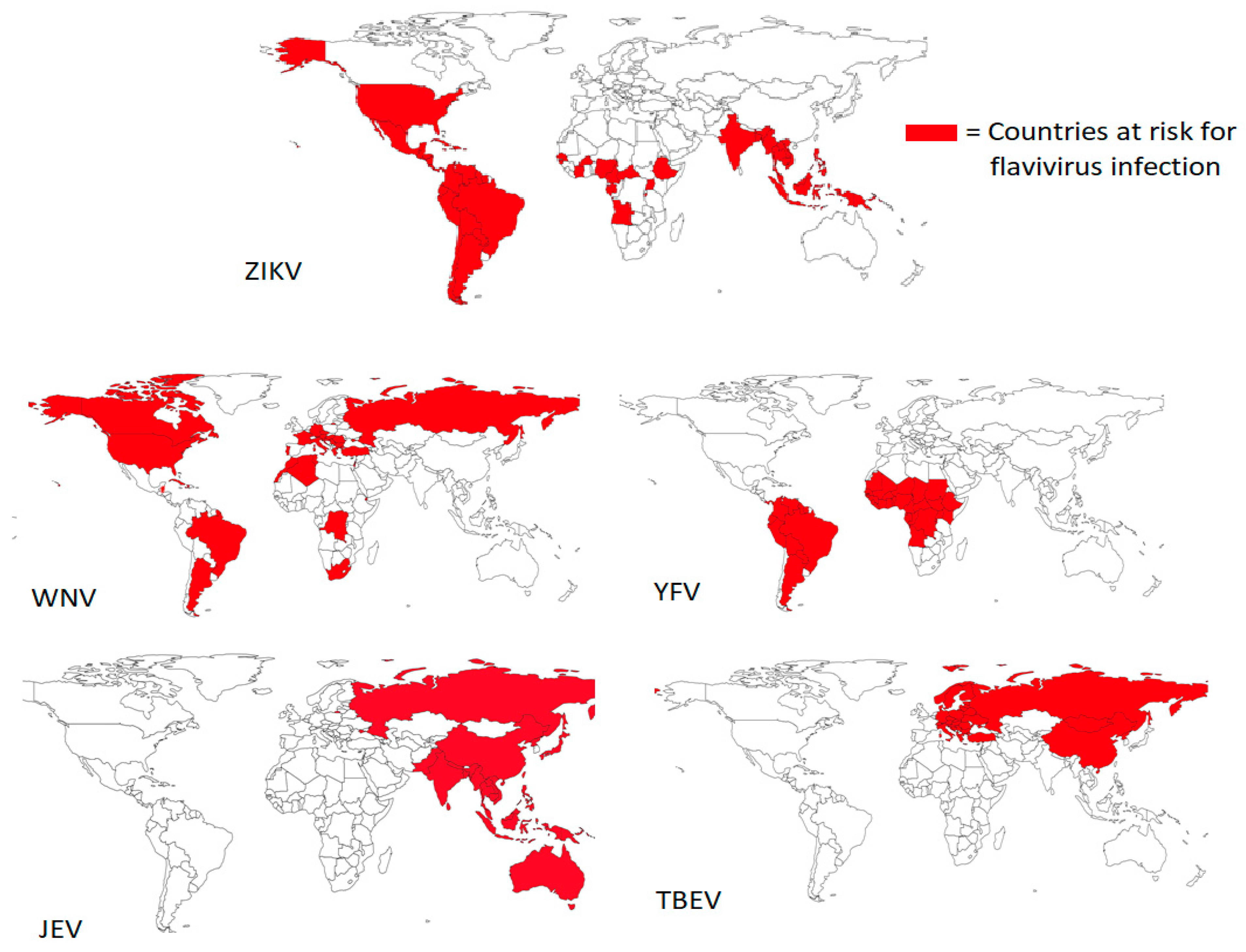
1.1. Epidemiology of Flaviviruses
1.2. Molecular Characterization and Life Cycle
1.3. Immune Response
1.4. Flavivirus Vaccines
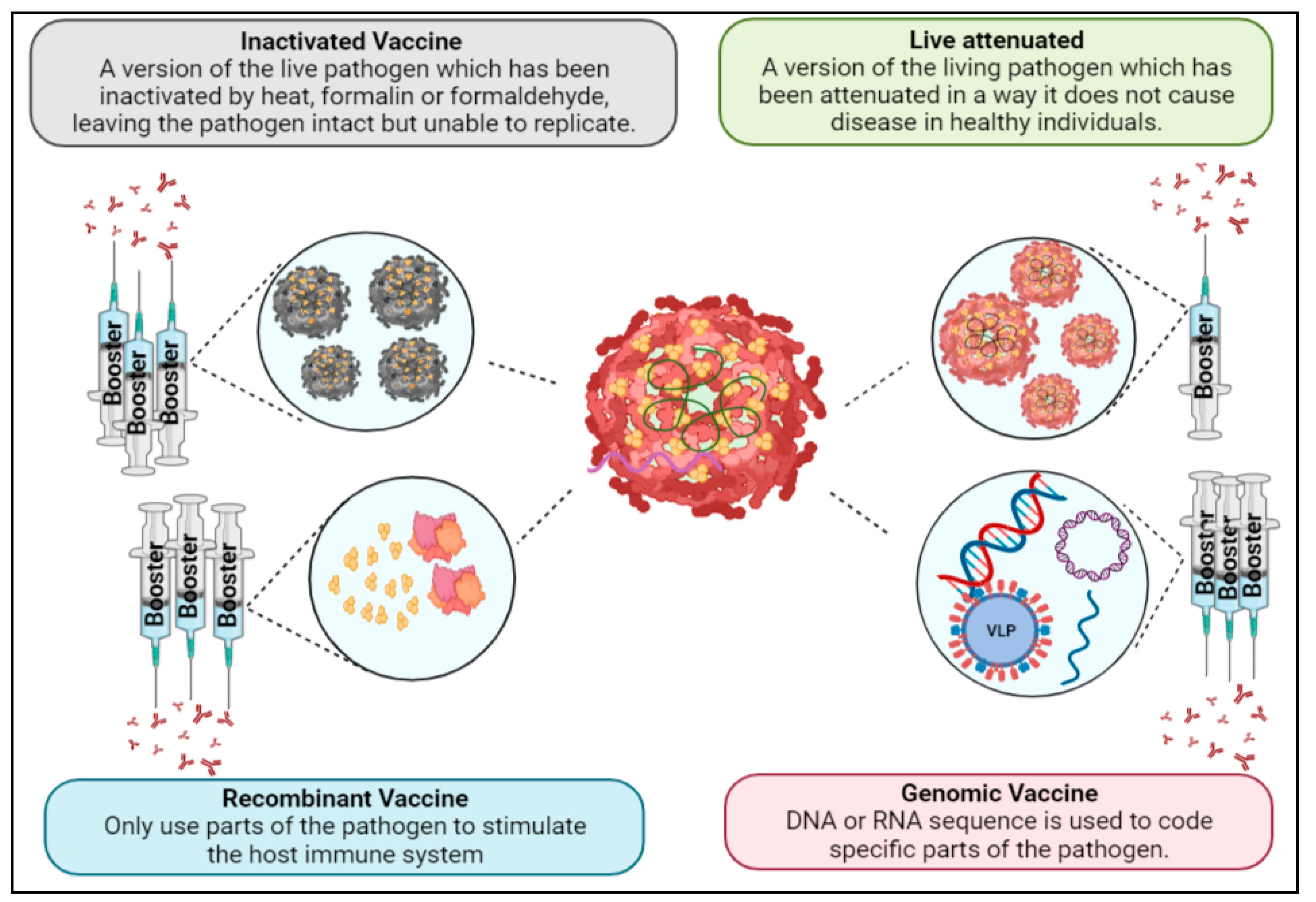
1.5. Vaccine Development
1.6. Live-Attenuated Vaccines
1.7. Inactivated Vaccines
1.8. Recombinant Vaccines
1.8.1. Subunit Vaccines
1.8.2. Virus-Like Particles
1.8.3. Viral Vector-Based Vaccines
1.8.4. Epitope-Based T-/B-Cell Vaccines
1.9. Genomic Vaccines
2. Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- MacLachlan, N.J.; Dubovi, E.J. Fenner’s Veterinary Virology, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 525–545. [Google Scholar]
- Kok, W.M. New developments in flavivirus drug discovery. Expert Opin. Drug Discov. 2016, 11, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dengue and Severe Dengue. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue (accessed on 15 December 2022).
- Dengue Vaccines: WHO Position Paper—September 2018 Weekly Epidemiological Record; WHO: Geneva, Switzerland, 2018; Volume 93, pp. 457–476.
- Halstead, S.B. Dengue Antibody-Dependent Enhancement: Knowns and Unknowns. Microbiol. Spectr. 2014, 2, 249–271. [Google Scholar] [CrossRef] [PubMed]
- Katzelnick, L.C.; Gresh, L.; Halloran, M.E.; Mercado, J.C.; Kuan, G.; Gordon, A.; Balmaseda, A.; Harris, E. Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358, 929–932. [Google Scholar] [CrossRef]
- Zorrilla, C.D.; Rivera-Viñas, J.I.; De La Vega-Pujols, A.; Garcia-Garcia, I.; Rabionet, S.E.; Mosquera, A.M. The Zika Virus Infection in Pregnancy: Review and Implications for Research and Care of Women and Infants in Affected Areas. Puerto Rico Health Sci. J. 2018, 37, S66–S72. [Google Scholar]
- WHO. Countries and Territories with Current or Previous Zika Virus Transmission. Updated February 2022. Available online: https://cdn.who.int/media/docs/default-source/documents/emergencies/zika/countries-with-zika-and-vectors-table_february-2022.pdf (accessed on 10 December 2022).
- Zika Epidemiology Update—February 2022. Available online: https://www.who.int/publications/m/item/zika-epidemiology-update---february-2022 (accessed on 8 February 2022).
- Dengue around the World—CDC. Available online: https://www.cdc.gov/dengue/areaswithrisk/around-the-world.html (accessed on 10 December 2022).
- Habarugira, G.; Suen, W.W.; Hobson-Peters, J.; Hall, R.A.; Bielefeldt-Ohmann, H. West Nile Virus: An Update on Pathobiology, Epidemiology, Diagnostics, Control and “One Health” Implications. Pathogens 2020, 9, 589. [Google Scholar] [CrossRef]
- West Nile Virus—CDC. Available online: https://www.cdc.gov/westnile/index.html (accessed on 15 December 2022).
- World Health Organization (2 September 2022). Disease Outbreak News; Yellow Fever in East, West, and Central Africa. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON405 (accessed on 10 December 2022).
- Centers for Disease Control and Prevention (2022) Geographic Distribution of Japanese Encephalitis Virus. Available online: https://www.cdc.gov/japaneseencephalitis/maps/index.html (accessed on 10 December 2022).
- Japanese Encephalitis Virus (JEV). Available online: https://www.health.gov.au/health-alerts/japanese-encephalitis-virus-jev/about#current-status (accessed on 10 December 2022).
- Tick-Borne Encephalitis. Available online: https://www.who.int/health-topics/tick-borne-encephalitis#tab=tab_1 (accessed on 15 December 2022).
- Radzišauskienė, D.; Urbonienė, J.; Kaubrys, G.; Andruškevičius, S.; Jatužis, D.; Matulytė, E.; Žvirblytė-Skrebutienė, K. The epidemiology, clinical presentation, and predictors of severe Tick-borne encephalitis in Lithuania, a highly endemic country: A retrospective study of 1040 patients. PLoS ONE 2020, 15, e0241587. [Google Scholar] [CrossRef] [PubMed]
- Douam, F.; Ploss, A. Yellow Fever Virus: Knowledge Gaps Impeding the Fight Against an Old Foe. Trends Microbiol. 2018, 26, 913–928. [Google Scholar] [CrossRef]
- Haslwanter, D.; Lasso, G.; Wec, A.Z.; Furtado, N.D.; Raphael, L.M.S.; Tse, A.L.; Sun, Y.; Stransky, S.; Pedreño-Lopez, N.; Correia, C.A.; et al. Genotype-specific features reduce the susceptibility of South American yellow fever virus strains to vaccine-induced antibodies. Cell Host Microbe 2022, 30, 248–259.e6. [Google Scholar] [CrossRef]
- Ricklin, M.E.; García-Nicolás, O.; Brechbühl, D.; Python, S.; Zumkehr, B.; Nougairede, A.; Charrel, R.N.; Posthaus, H.; Oevermann, A.; Summerfield, A. Vector-free transmission and persistence of Japanese encephalitis virus in pigs. Nat. Commun. 2016, 7, 10832. [Google Scholar] [CrossRef]
- Caldwell, J.P.; Chen, L.H.; Hamer, D.H. Evolving Epidemiology of Japanese Encephalitis: Implications for Vaccination. Curr. Infect. Dis. Rep. 2018, 20, 30. [Google Scholar] [CrossRef]
- Weekly Updates: 2022 West Nile Virus Transmission Season. Available online: https://www.ecdc.europa.eu/en/west-nile-fever/surveillance-and-disease-data/disease-data-ecdc (accessed on 15 December 2022).
- European Centre for Disease Prevention and Control. Tick-Borne Encephalitis. In ECDC. Annual Epidemiological Report for 2022; ECDC: Stockholm, Sweden, 2022. [Google Scholar]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef]
- Barrows, N.J.; Campos, R.K.; Liao, K.-C.; Prasanth, K.R.; Soto-Acosta, R.; Yeh, S.-C.; Schott-Lerner, G.; Pompon, J.; Sessions, O.M.; Bradrick, S.S.; et al. Biochemistry and Molecular Biology of Flaviviruses. Chem. Rev. 2018, 118, 4448–4482. [Google Scholar] [CrossRef]
- Pierson, T.C.; Kielian, M. Flaviviruses: Braking the entering. Curr. Opin. Virol. 2013, 3, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, M.D.; Mazzon, M.; Jacobs, M.; Amara, A. Pathogenesis of Flavivirus Infections: Using and Abusing the Host Cell. Cell Host Microbe 2009, 5, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.A.; Stiasny, K.; Vaney, M.C.; Dellarole, M.; Heinz, F.X. The bright and the dark side of human antibody responses to flaviviruses: Lessons for vaccine design. EMBO Rep. 2018, 19, 206–224. [Google Scholar] [CrossRef] [PubMed]
- Slon Campos, J.L.; Mongkolsapaya, J.; Screaton, G.R. The immune response against flaviviruses. Nat. Immunol. 2018, 19, 1189–1198. [Google Scholar] [CrossRef]
- de Alwis, R.; Smith, S.A.; Olivarez, N.P.; Messer, W.B.; Huynh, J.P.; Wahala, W.M.; White, L.J.; Diamond, M.S.; Baric, R.S.; Crowe, J.E., Jr.; et al. Identification of human neutralizing antibodies that bind to complex epitopes on dengue virions. Proc. Natl. Acad. Sci. USA 2012, 109, 7439–7444. [Google Scholar] [CrossRef]
- Ooi, J.S.G.; Lok, S.-M. How NS1 Antibodies Prevent Severe Flavivirus Disease. Trends Biochem. Sci. 2021, 46, 519–521. [Google Scholar] [CrossRef]
- Falconar, A.K. The dengue virus nonstructural-1 protein (NS1) generates antibodies to common epitopes on human blood clotting, integrin/adhesin proteins and binds to human endothelial cells: Potential implications in haemorrhagic fever pathogenesis. Arch. Virol. 1997, 142, 897–916. [Google Scholar] [CrossRef]
- Beatty, P.R.; Puerta-Guardo, H.; Killingbeck, S.S.; Glasner, D.R.; Hopkins, K.; Harris, E. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci. Transl. Med. 2015, 7, 304ra141. [Google Scholar] [CrossRef] [PubMed]
- Martina, B.E.; Koraka, P.; Osterhaus, A.D. Dengue Virus Pathogenesis: An Integrated View. Clin. Microbiol. Rev. 2009, 22, 564–581. [Google Scholar] [CrossRef]
- Ishikawa, T.; Yamanaka, A.; Konishi, E. A review of successful flavivirus vaccines and the problems with those flaviviruses for which vaccines are not yet available. Vaccine 2014, 32, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Rattan, A.; Richards, K.A.; Knowlden, Z.A.G.; Sant, A.J. Protein Vaccination Directs the CD4 + T Cell Response toward Shared Protective Epitopes That Can Be Recalled after Influenza Virus Infection. J. Virol. 2019, 93, e00947-19. [Google Scholar] [CrossRef]
- Sharma, G.; Rive, C.M.; Holt, R.A. Rapid selection and identification of functional CD8+ T cell epitopes from large peptide-coding libraries. Nat. Commun. 2019, 10, 4553. [Google Scholar] [CrossRef]
- Kräutler, N.J.; Suan, D.; Butt, D.; Bourne, K.; Hermes, J.R.; Chan, T.D.; Sundling, C.; Kaplan, W.; Schofield, P.; Jackson, J.; et al. Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J. Exp. Med. 2017, 214, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.D.; Okada, T.; Cyster, J.G. Germinal-Center Organization and Cellular Dynamics. Immunity 2007, 27, 190–202. [Google Scholar] [CrossRef]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2021, 21, 83–100. [Google Scholar] [CrossRef]
- Dai, X.; Xiong, Y.; Li, N.; Jian, C. Vaccine Types. 2019. Vaccines—The History and Future. IntechOpen. Available online: https://www.intechopen.com/chapters/undefined/state.item.id (accessed on 21 December 2022).
- Approved Vaccine Products|FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/approved-vaccine-products (accessed on 31 January 2023).
- WHO Guidelines on Non-Clinical Evaluation of Vaccines, Annex 1, TRS No 927. Available online: https://www.who.int/publications/m/item/nonclinical-evaluation-of-vaccines-annex-1-trs-no-927 (accessed on 10 December 2022).
- Lebrón, J.A.; Wolf, J.J.; Kaplanski, C.V.; Ledwith, B.J. Ensuring the quality, potency and safety of vaccines during preclinical development. Expert Rev. Vaccines 2005, 4, 855–866. [Google Scholar] [CrossRef]
- World Health Organization (2003) Guidelines on Nonclinical Evaluation of Vaccines. World Health Organization, Geneva. Available online: https://www.impfkritik.de/upload/pdf/wirksamkeitsnachweis/WHO-2003.pdf (accessed on 31 January 2023).
- World Health Organization. Guidelines for Good Clinical Practice (GCP) for Trials on Pharmaceutical Products; Technical Report Series; WHO: Geneva, Switzerland, 1995; Volume 850, pp. 97–137. [Google Scholar]
- Vaccine Development—101|FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/development-approval-process-cber/vaccine-development-101 (accessed on 31 January 2023).
- Umscheid, C.A.; Margolis, D.J.; Grossman, C.E. Key Concepts of Clinical Trials: A Narrative Review. Postgrad. Med. 2011, 123, 194–204. [Google Scholar] [CrossRef]
- Minor, P.D. Live attenuated vaccines: Historical successes and current challenges. Virology 2015, 479–480, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Marohn, M.E.; Barry, E.M. Live attenuated tularemia vaccines: Recent developments and future goals. Vaccine 2013, 31, 3485–3491. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.D.T. Yellow fever live attenuated vaccine: A very successful live attenuated vaccine but still we have problems controlling the disease. Vaccine 2017, 35, 5951–5955. [Google Scholar] [CrossRef] [PubMed]
- Lobigs, M.; Larena, M.; Alsharifi, M.; Lee, E.; Pavy, M. Live Chimeric and Inactivated Japanese Encephalitis Virus Vaccines Differ in Their Cross-Protective Values against Murray Valley Encephalitis Virus. J. Virol. 2009, 83, 2436–2445. [Google Scholar] [CrossRef]
- Hanley, K.A. The Double-Edged Sword: How Evolution Can Make or Break a Live-Attenuated Virus Vaccine. Evolution 2011, 4, 635–643. [Google Scholar] [CrossRef]
- Coleman, J.R.; Papamichail, D.; Skiena, S.; Futcher, B.; Wimmer, E.; Mueller, S. Virus attenuation by genome-scale changes in codon pair bias. Science 2008, 320, 1784–1787. [Google Scholar] [CrossRef]
- Lauring, A.S.; Jones, J.O.; Andino, R. Rationalizing the development of live attenuated virus vaccines. Nat. Biotechnol. 2010, 28, 573–579. [Google Scholar] [CrossRef]
- Collins, N.D.; Barrett, A.D.T. Live Attenuated Yellow Fever 17D Vaccine: A Legacy Vaccine Still Controlling Outbreaks In Modern Day. Curr. Infect. Dis. Rep. 2017, 19, 14. [Google Scholar] [CrossRef]
- Thomas, R.E. Yellow fever vaccine-associated viscerotropic disease: Current perspectives. Drug Des. Dev. Ther. 2016, 10, 3345–3353. [Google Scholar] [CrossRef]
- Yang, D.; Li, X.-F.; Ye, Q.; Wang, H.-J.; Deng, Y.-Q.; Zhu, S.-Y.; Zhang, Y.; Li, S.-H.; Qin, C.-F. Characterization of live-attenuated Japanese encephalitis vaccine virus SA14-14-2. Vaccine 2014, 32, 2675–2681. [Google Scholar] [CrossRef]
- Appaiahgari, M.B.; Vrati, S. IMOJEV®: A Yellow fever virus-based novel Japanese encephalitis vaccine. Expert Rev. Vaccines 2010, 9, 1371–1384. [Google Scholar] [CrossRef] [PubMed]
- Vannice, K.S.; Roehrig, J.T.; Hombach, J. Next generation dengue vaccines: A review of the preclinical development pipeline. Vaccine 2015, 33, 7091–7099. [Google Scholar] [CrossRef] [PubMed]
- Henein, S.; Swanstrom, J.; Byers, A.M.; Moser, J.M.; Shaik, S.F.; Bonaparte, M.; Jackson, N.; Guy, B.; Baric, R.; de Silva, A.M. Dissecting antibodies induced by a chimeric yellow fever-dengue, live-attenuated, tetravalent dengue vaccine (CYD-TDV) in naïve and dengue exposed individuals. J. Infect. Dis. 2017, 215, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Yoon, I.-K. A review of Dengvaxia®: Development to deployment. Hum. Vaccines Immunother. 2019, 15, 2295–2314. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. Dengvaxia sensitizes seronegatives to vaccine enhanced disease regardless of age. Vaccine 2017, 35, 6355–6358. [Google Scholar] [CrossRef]
- Precioso, A.R.; Palacios, R.; Thomé, B.; Mondini, G.; Braga, P.; Kalil, J. Clinical evaluation strategies for a live attenuated tetravalent dengue vaccine. Vaccine 2015, 33, 7121–7125. [Google Scholar] [CrossRef] [PubMed]
- Osorio, J.E.; Wallace, D.; Stinchcomb, D.T. A recombinant, chimeric tetravalent dengue vaccine candidate based on a dengue virus serotype 2 backbone. Expert Rev. Vaccines 2016, 15, 497–508. [Google Scholar] [CrossRef]
- Sáez-Llorens, X.; Tricou, V.; Yu, D.; Rivera, L.; Tuboi, S.; Garbes, P.; Borkowski, A.; Wallace, D. Safety and immunogenicity of one versus two doses of Takeda’s tetravalent dengue vaccine in children in Asia and Latin America: Interim results from a phase 2, randomised, placebo-controlled study. Lancet Infect. Dis. 2017, 17, 615–625. [Google Scholar] [CrossRef]
- Hadinegoro, S.R.; Arredondo-García, J.L.; Capeding, M.R.; Deseda, C.; Chotpitayasunondh, T.; Dietze, R.; Muhammad Ismail, H.I.; Reynales, H.; Limkittikul, K.; Rivera-Medina, D.M.; et al. CYD-TDV Dengue Vaccine Working Group. Efficacy and Long-Term Safety of a Dengue Vaccine in Regions of Endemic Disease. N. Engl. J. Med. 2015, 373, 1195–1206. [Google Scholar] [CrossRef]
- Pletnev, A.G.; Claire, M.S.; Elkins, R.; Speicher, J.; Murphy, B.R.; Chanock, R.M. Molecularly engineered live-attenuated chimeric West Nile/dengue virus vaccines protect rhesus monkeys from West Nile virus. Virology 2003, 314, 190–195. [Google Scholar] [CrossRef]
- Monath, T.P.; Liu, J.; Kanesa-Thasan, N.; Myers, G.A.; Nichols, R.; Deary, A.; McCarthy, K.; Johnson, C.; Ermak, T.; Shin, S.; et al. A live, attenuated recombinant West Nile virus vaccine. Proc. Natl. Acad. Sci. USA 2006, 103, 6694–6699. [Google Scholar] [CrossRef] [PubMed]
- Delrue, I.; Verzele, D.; Madder, A.; Nauwynck, H.J. Inactivated virus vaccines from chemistry to prophylaxis: Merits, risks and challenges. Expert Rev. Vaccines 2012, 11, 695–719. [Google Scholar] [CrossRef] [PubMed]
- Brown, F. Review of accidents caused by incomplete inactivation of viruses. Dev. Biol. Stand. 1993, 81, 103–107. [Google Scholar] [PubMed]
- Reisler, R.B.; Danner, D.K.; Gibbs, P.H. Immunogenicity of an inactivated Japanese encephalitis vaccine (JE-VAX) in humans over 20 years at USAMRIID: Using PRNT50 as an endpoint for immunogenicity. Vaccine 2010, 28, 2436–2441. [Google Scholar] [CrossRef]
- Lindsey, N.P.; Staples, J.E.; Jones, J.F.; Sejvar, J.J.; Griggs, A.; Iskander, J.; Miller, E.R.; Fischer, M. Adverse event reports following Japanese encephalitis vaccination in the United States, 1999–2009. Vaccine 2010, 29, 58–64. [Google Scholar] [CrossRef]
- Mansfield, K.L.; Johnson, N.; Phipps, L.P.; Stephenson, J.R.; Fooks, A.R.; Solomon, T. Tick-borne encephalitis virus—A review of an emerging zoonosis. J. Gen. Virol. 2009, 90, 1781–1794. [Google Scholar] [CrossRef]
- Loew-Baselli, A.; Poellabauer, E.M.; Pavlova, B.G.; Fritsch, S.; Firth, C.; Petermann, R.; Barrett, P.N.; Ehrlich, H.J. Prevention of tick-borne encephalitis by FSME-IMMUN® vaccines: Review of a clinical development programme. Vaccine 2011, 29, 7307–7319. [Google Scholar] [CrossRef]
- Joachim, H.; Alan, D.T. Barrett, Herwig Kollaritsch, Tickborne Encephalitis Vaccines. In Plotkin’s Vaccines, 7th ed.; Plotkin, S.A., Orenstein, W.A., Offit, P.A., Edwards, K.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1080–1094.e5. [Google Scholar]
- Fernandez, S.; Thomas, S.J.; De La Barrera, R.; Im-Erbsin, R.; Jarman, R.G.; Baras, B.; Toussaint, J.-F.; Mossman, S.; Innis, B.; Schmidt, A.; et al. An Adjuvanted, Tetravalent Dengue Virus Purified Inactivated Vaccine Candidate Induces Long-Lasting and Protective Antibody Responses Against Dengue Challenge in Rhesus Macaques. Am. J. Trop. Med. Hyg. 2015, 92, 698–708. [Google Scholar] [CrossRef]
- Modjarrad, K.; Lin, L.; George, S.L.; Stephenson, K.E.; Eckels, K.H.; De La Barrera, R.A.; Jarman, R.G.; Sondergaard, E.; Tennant, J.; Ansel, J.L.; et al. Preliminary aggregate safety and immunogenicity results from three trials of a purified inactivated Zika virus vaccine candidate: Phase 1, randomised, double-blind, placebo-controlled clinical trials. Lancet 2018, 391, 563–571. [Google Scholar] [CrossRef]
- Han, H.-H.; Diaz, C.; Acosta, C.J.; Liu, M.; Borkowski, A. Safety and immunogenicity of a purified inactivated Zika virus vaccine candidate in healthy adults: An observer-blind, randomised, phase 1 trial. Lancet Infect. Dis. 2021, 21, 1282–1292. [Google Scholar] [CrossRef]
- Baldwin, W.R.; Livengood, J.A.; Giebler, H.A.; Stovall, J.L.; Boroughs, K.L.; Sonnberg, S.; Bohning, K.J.; Dietrich, E.A.; Ong, Y.T.; Danh, H.K.; et al. Purified Inactivated Zika Vaccine Candidates Afford Protection against Lethal Challenge in Mice. Sci. Rep. 2018, 8, 16509. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P.; Fowler, E.; Johnson, C.T.; Balser, J.; Morin, M.J.; Sisti, M.; Trent, D.W. An Inactivated Cell-Culture Vaccine against Yellow Fever. N. Engl. J. Med. 2011, 364, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P.; Lee, C.K.; Julander, J.G.; Brown, A.; Beasley, D.W.; Watts, D.M.; Hayman, E.; Guertin, P.; Makowiecki, J.; Crowell, J.; et al. Inactivated yellow fever 17D vaccine: Development and nonclinical safety, immunogenicity and protective activity. Vaccine 2010, 28, 3827–3840. [Google Scholar] [CrossRef]
- Moyle, P.M.; Toth, I. Modern Subunit Vaccines: Development, Components, and Research Opportunities. Chemmedchem 2013, 8, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Manoff, S.B.; Sausser, M.; Falk Russell, A.; Martin, J.; Radley, D.; Hyatt, D.; Roberts, C.; Lickliter, J.; Krishnarajah, J.; Bett, A.; et al. Immunogenicity and safety of an investigational tetravalent recombinant subunit vaccine for dengue: Results of a Phase I randomized clinical trial in flavivirus-naïve adults. Hum. Vaccines Immunother. 2019, 15, 2195–2204. [Google Scholar] [CrossRef]
- Lieberman, M.M.; Nerurkar, V.R.; Luo, H.; Cropp, B.; Carrion, R.; de la Garza, M.; Coller, B.-A.; Clements, D.; Ogata, S.; Wong, T.; et al. Immunogenicity and Protective Efficacy of a Recombinant Subunit West Nile Virus Vaccine in Rhesus Monkeys. Clin. Vaccine Immunol. 2009, 16, 1332–1337. [Google Scholar] [CrossRef]
- Dai, S.; Wang, H.; Deng, F. Advances and challenges in enveloped virus-like particle (VLP)-based vaccines. J. Immunol. Sci. 2018, 2, 36–41. [Google Scholar]
- Krol, E.; Brzuska, G.; Szewczyk, B. Production and Biomedical Application of Flavivirus-like Particles. Trends Biotechnol. 2019, 37, 1202–1216. [Google Scholar] [CrossRef]
- Fuenmayor, J.; Gòdia, F.; Cervera, L. Production of virus-like particles for vaccines. New Biotechnol. 2017, 39 Pt B, 174–180. [Google Scholar] [CrossRef]
- Cabral-Miranda, G.; Lim, S.M.; Mohsen, M.O.; Pobelov, I.V.; Roesti, E.S.; Heath, M.D.; Skinner, M.A.; Kramer, M.F.; Martina, B.E.E.; Bachmann, M.F. Zika Virus-Derived E-DIII Protein Displayed on Immunologically Optimized VLPs Induces Neutralizing Antibodies without Causing Enhancement of Dengue Virus Infection. Vaccines 2019, 7, 72. [Google Scholar] [CrossRef]
- Gil, L.; Cobas, K.; Lazo, L.; Marcos, E.; Hernández, L.; Suzarte, E.; Izquierdo, A.; Valdés, I.; Blanco, A.; Puentes, P.; et al. A Tetravalent Formulation Based on Recombinant Nucleocapsid-like Particles from Dengue Viruses Induces a Functional Immune Response in Mice and Monkeys. J. Immunol. 2016, 197, 3597–3606. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Juárez, E.; Pando-Robles, V.; Palomares, L.A. Flavivirus vaccines: Virus-like particles and single-round infectious particles as promising alternatives. Vaccine 2021, 39, 6990–7000. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Jasny, E.; Schmidt, K.E.; Petsch, B. New Vaccine Technologies to Combat Outbreak Situations. Front. Immunol. 2018, 9, 1963. [Google Scholar] [CrossRef]
- van den Baldo, A.; Akker, E.; Bergmans, H.; Lim, F.; Pauwels, K. General Considerations on the Biosafety of Virus-derived Vectors Used in Gene Therapy and Vaccination. Curr. Gene Ther. 2013, 13, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Condit, R.C.; Williamson, A.-L.; Sheets, R.; Seligman, S.J.; Monath, T.P.; Excler, J.-L.; Gurwith, M.; Bok, K.; Robertson, J.S.; Kim, D.; et al. Unique safety issues associated with virus-vectored vaccines: Potential for and theoretical consequences of recombination with wild type virus strains. Vaccine 2016, 34, 6610–6616. [Google Scholar] [CrossRef] [PubMed]
- Nürnberger, C.; Bodmer, B.S.; Fiedler, A.H.; Gabriel, G.; Mühlebach, M.D. A Measles Virus-Based Vaccine Candidate Mediates Protection against Zika Virus in an Allogeneic Mouse Pregnancy Model. J. Virol. 2019, 93, e01485-18. [Google Scholar] [CrossRef]
- Oyarzún, P.; Kobe, B. Recombinant and epitope-based vaccines on the road to the market and implications for vaccine design and production. Hum. Vaccines Immunother. 2016, 12, 763–767. [Google Scholar] [CrossRef]
- Li, S.; Peng, L.; Zhao, W.; Zhong, H.; Zhang, F.; Yan, Z.; Cao, H. Synthetic peptides containing B- and T-cell epitope of dengue virus-2 E domain III provoked B- and T-cell responses. Vaccine 2011, 29, 3695–3702. [Google Scholar] [CrossRef] [PubMed]
- Wollner, C.J.; Richner, M.; Hassert, M.A.; Pinto, A.K.; Brien, J.D.; Richner, J.M. A Dengue Virus Serotype 1 mRNA-LNP Vaccine Elicits Protective Immune Responses. J. Virol. 2021, 95, e02482-20. [Google Scholar] [CrossRef]
- Lin, H.-H.; Yip, B.-S.; Huang, L.-M.; Wu, S.-C. Zika virus structural biology and progress in vaccine development. Biotechnol. Adv. 2018, 36, 47–53. [Google Scholar] [CrossRef]
- Lei, Y.; Zhao, F.; Shao, J.; Li, Y.; Li, S.; Chang, H.; Zhang, Y. Application of built-in adjuvants for epitope-based vaccines. PeerJ 2019, 6, e6185. [Google Scholar] [CrossRef] [PubMed]
- Hobernik, D.; Bros, M. DNA Vaccines-How Far from Clinical Use? Int. J. Mol. Sci. 2018, 19, 3605. [Google Scholar] [CrossRef] [PubMed]
- Danko, J.R.; Beckett, C.G.; Porter, K.R. Development of dengue DNA vaccines. Vaccine 2011, 29, 7261–7266. [Google Scholar] [CrossRef]
- Pushparajah, D.; Jimenez, S.; Wong, S.; Alattas, H.; Nafissi, N.; Slavcev, R.A. Advances in gene-based vaccine platforms to address the COVID-19 pandemic. Adv. Drug Deliv. Rev. 2021, 170, 113–141. [Google Scholar] [CrossRef] [PubMed]
- Klinman, D.M.; Takeno, M.; Ichino, M.; Gu, M.; Yamshchikov, G.; Mor, G.; Conover, J. DNA vaccines: Safety and efficacy issues. Springer Semin. Immunopathol. 1997, 19, 245–256. [Google Scholar] [CrossRef]
- Sullivan, S.M.; Doukas, J.; Hartikka, J.; Smith, L.; Rolland, A. Vaxfectin: A versatile adjuvant for plasmid DNA- and protein-based vaccines. Expert Opin. Drug Deliv. 2010, 7, 1433–1446. [Google Scholar] [CrossRef]
- Danko, J.R.; Kochel, T.; Teneza-Mora, N.; Luke, T.C.; Raviprakash, K.; Sun, P.; Simmons, M.; Moon, J.E.; De La Barrera, R.; Martinez, L.J.; et al. Safety and Immunogenicity of a Tetravalent Dengue DNA Vaccine Administered with a Cationic Lipid-Based Adjuvant in a Phase 1 Clinical Trial. Am. J. Trop. Med. Hyg. 2018, 98, 849–856. [Google Scholar] [CrossRef]
- Ledgerwood, J.E.; Pierson, T.C.; Hubka, S.A.; Desai, N.; Rucker, S.; Gordon, I.J.; Enama, M.E.; Nelson, S.; Nason, M.; Gu, W.; et al. A West Nile Virus DNA Vaccine Utilizing a Modified Promoter Induces Neutralizing Antibody in Younger and Older Healthy Adults in a Phase I Clinical Trial. J. Infect. Dis. 2011, 203, 1396–1404. [Google Scholar] [CrossRef]
- Dowd, K.A.; Ko, S.Y.; Morabito, K.M.; Yang, E.S.; Pelc, R.S.; DeMaso, C.R.; Castilho, L.R.; Abbink, P.; Boyd, M.; Nityanandam, R.; et al. Rapid development of a DNA vaccine for Zika virus. Science 2016, 354, 237–240. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. COVE Study Group. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Thomas, S.J.; Moreira, E.D., Jr.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Polack, F.P.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months. N. Engl. J. Med. 2021, 385, 1761–1773. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, J.; Liu, M. Ethical issues for vaccines and immunization. Nat. Rev. Immunol. 2002, 2, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Castanha, P.M.S.; Nascimento, E.J.M.; Braga, C.; Cordeiro, M.T.; Carvalho, O.V.; Mendonça, L.R. Dengue virus (DENV)-specific antibodies enhance Brazilian Zika virus (ZIKV) infection. J. Infect. Dis. 2017, 215, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Gershman, M.D.; Staples, J.E.; Bentsi-Enchill, A.D.; Breugelmans, J.G.; Brito, G.S.; Camacho, L.A.B.; Cottin, P.; Domingo, C.; Durbin, A.; Gascon, J.; et al. Viscerotropic disease: Case definition and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine 2012, 30, 5038–5058. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.; Miller, C.; Catalan, J.; Myers, G.A.; Ratterree, M.S.; Trent, D.W.; Monath, T.P. ChimeriVax-West Nile virus live-attenuated vaccine: Preclinical evaluation of safety, immunogenicity, and efficacy. J. Virol. 2004, 78, 12497–12507. [Google Scholar] [CrossRef]
- Kwek, S.S.; Watanabe, S.; Chan, K.R.; Ong, E.Z.; Tan, H.C.; Ng, W.C.; Nguyen, M.T.X.; Gan, E.S.; Zhang, S.L.; Chan, K.W.K.; et al. A systematic approach to the development of a safe live attenuated Zika vaccine. Nat. Commun. 2018, 9, 1031. [Google Scholar] [CrossRef]
- Schmidt, A.C.; Lin, L.; Martinez, L.J.; Ruck, R.C.; Eckels, K.H.; Collard, A.; De La Barrera, R.; Paolino, K.M.; Toussaint, J.-F.; Lepine, E.; et al. Phase 1 Randomized Study of a Tetravalent Dengue Purified Inactivated Vaccine in Healthy Adults in the United States. Am. J. Trop. Med. Hyg. 2017, 96, 1325–1337. [Google Scholar] [CrossRef]
- Nascimento, I.P.; Leite, L.C. Recombinant vaccines and the development of new vaccine strategies. Braz. J. Med. Biol. Res. 2012, 45, 1102–1111. [Google Scholar] [CrossRef]
- Schiller, J.T.; Lowy, D.R. Raising Expectations for Subunit Vaccine. J. Infect. Dis. 2015, 211, 1373–1375. [Google Scholar] [CrossRef]
- Lin, H.-H.; Yang, S.-P.; Tsai, M.-J.; Lin, G.-C.; Wu, H.-C.; Wu, S.-C. Dengue and Zika Virus Domain III-Flagellin Fusion and Glycan-Masking E Antigen for Prime-Boost Immunization. Theranostics 2019, 9, 4811–4826. [Google Scholar] [CrossRef]
- McDonald, W.F.; Huleatt, J.W.; Foellmer, H.G.; Hewitt, D.D.; Tang, J.; Desai, P.; Price, A.E.; Jacobs, A.; Takahashi, V.N.; Huang, Y.; et al. A West Nile Virus Recombinant Protein Vaccine That Coactivates Innate and Adaptive Immunity. J. Infect. Dis. 2007, 195, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Song, L.; Beasley, D.W.C.; Putnak, R.; Parent, J.; Misczak, J.; Li, H.; Reiserova, L.; Liu, X.; Tian, H.; et al. Immunogenicity and Efficacy of Flagellin-Envelope Fusion Dengue Vaccines in Mice and Monkeys. Clin. Vaccine Immunol. 2015, 22, 516–525. [Google Scholar] [CrossRef]
- Treanor, J.J.; Taylor, D.N.; Tussey, L.; Hay, C.; Nolan, C.; Fitzgerald, T.; Liu, G.; Kavita, U.; Song, L.; Dark, I.; et al. Safety and immunogenicity of a recombinant hemagglutinin influenza–flagellin fusion vaccine (VAX125) in healthy young adults. Vaccine 2010, 28, 8268–8274. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.N.; Treanor, J.J.; Strout, C.; Johnson, C.; Fitzgerald, T.; Kavita, U.; Ozer, K.; Tussey, L.; Shaw, A. Induction of a potent immune response in the elderly using the TLR-5 agonist, flagellin, with a recombinant hemagglutinin influenza–flagellin fusion vaccine (VAX125, STF2.HA1 SI). Vaccine 2011, 29, 4897–4902. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major findings and recent advances in virus–like particle (VLP)-based vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef]
- Pardy, R.D.; Richer, M.J. Protective to a T: The Role of T Cells during Zika Virus Infection. Cells 2019, 8, 820. [Google Scholar] [CrossRef]
- Williams, M.; Ewing, D.; Blevins, M.; Sun, P.; Sundaram, A.K.; Raviprakash, K.S.; Porter, K.R.; Sanders, J.W. Enhanced immunogenicity and protective efficacy of a tetravalent dengue DNA vaccine using electroporation and intradermal delivery. Vaccine 2019, 37, 4444–4453. [Google Scholar] [CrossRef]
- Grunwald, T.; Ulbert, S. Improvement of DNA vaccination by adjuvants and sophisticated delivery devices: Vaccine-platforms for the battle against infectious diseases. Clin. Exp. Vaccine Res. 2015, 4, 1–10. [Google Scholar] [CrossRef]
- Suschak, J.; Williams, J.A.; Schmaljohn, C.S. Advancements in DNA vaccine vectors, non-mechanical delivery methods, and molecular adjuvants to increase immunogenicity. Hum. Vaccines Immunother. 2017, 13, 2837–2848. [Google Scholar] [CrossRef]
- Farris, E.; Brown, D.M.; Ramer-Tait, A.E.; Pannier, A.K. Micro- and nanoparticulates for DNA vaccine delivery. Exp. Biol. Med. 2016, 241, 919–929. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flavivirus | Mild Symptoms | Severe Symptoms | Complications | Regions | Vectors |
|---|---|---|---|---|---|
| DENV | Fever, nausea, vomiting | Hemorrhagic fever, hematemesis | Cardiomyopathy, shock syndrome | The Americas, Africa, the Middle East and the Pacific Island | Ae. aegypti, Ae. albopictus |
| ZIKV | Fever, rash, headache | Long-term neurological complications | Birth defect, in rare cases GBS or cerebral edema | The Americas, Africa, Asia and the Pacific Island | Ae. aegypti, Ae. albopictus |
| WNV | Fever, headache, body aches | High fever, coma, tremors, convulsions, vision loss, numbness and paralysis, myalgia | Encephalitis, meningitis | Africa, Europe the Middle East, North America and west Asia | Culex pipiens |
| YFV | Fever, chills, severe headache | High fever, jaundice, bleeding | Shock, organ failure | Africa, central and south America | Aedes aegypti, Aedes africanus, Haemagogus spp, Sabethes spp. |
| TBEV | Fever, malaise, anorexia, muscle aches, headache, nausea and/or vomiting | Drowsiness, confusion, sensory disturbances, paralysis | Encephalitis, meningitis, meningoencephalitis | Europe and Asia | Ixodidae ricinus |
| JEV | Fever, headache, vomiting | Neurologic symptoms, seizures | Encephalitis, neurologic, cognitive or psychiatric symptoms after disease | Southeast Asia and the Pacific Island | Culex tritaeniorhynchus |
| Name | Vaccine Type | Virus | Manufacturer |
|---|---|---|---|
| CYD-TVD/Dengvaxia | Chimeric live-attenuated | DENV | Sanofi Pasteur |
| IC51/IXIARO | Inactive | JEV | WRAIR |
| JE-VAX | Inactive | JEV | The Research Foundation for Microbial Disease of Osaka University |
| SA 14-14-2 | Live-attenuated | JEV | Chengdu Institute of Biological Product |
| IMOJEV/JE-CV | Live-attenuated | JEV | Sanofi Pasture |
| TBE-Moscow | Inactive | TBEV | Chumakov Institute of Poliomyelitis and Viral Encephalitides |
| EnceVir | Inactive | TBEV | Microgen |
| FSME-IMMUN | Inactive | TBEV | Baxter |
| Encepur | Inactive | TBEV | Novartis |
| YFV-17DD | Live-attenuated | YFV | Bio-Manguinhos (Fiocruz) |
| YFV-17D-204 | Live-attenuated | YFV | Sanofi Pasteur Institute Chiron/Novartis |
| YFV-17D-213 | Live-attenuated | YFV | Federal State Unitary Enterprise of Chumakov Institute |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutta, S.K.; Langenburg, T. A Perspective on Current Flavivirus Vaccine Development: A Brief Review. Viruses 2023, 15, 860. https://doi.org/10.3390/v15040860
Dutta SK, Langenburg T. A Perspective on Current Flavivirus Vaccine Development: A Brief Review. Viruses. 2023; 15(4):860. https://doi.org/10.3390/v15040860
Chicago/Turabian StyleDutta, Sudip Kumar, and Thomas Langenburg. 2023. "A Perspective on Current Flavivirus Vaccine Development: A Brief Review" Viruses 15, no. 4: 860. https://doi.org/10.3390/v15040860
APA StyleDutta, S. K., & Langenburg, T. (2023). A Perspective on Current Flavivirus Vaccine Development: A Brief Review. Viruses, 15(4), 860. https://doi.org/10.3390/v15040860