
The Complex Role of HBeAg and Its Precursors in the Pathway to Hepatocellular Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- persist in liver tissue, increasing chronic oxidative damage in hepatocytes, immune-mediated inflammation of the liver, and development of HCC [12].
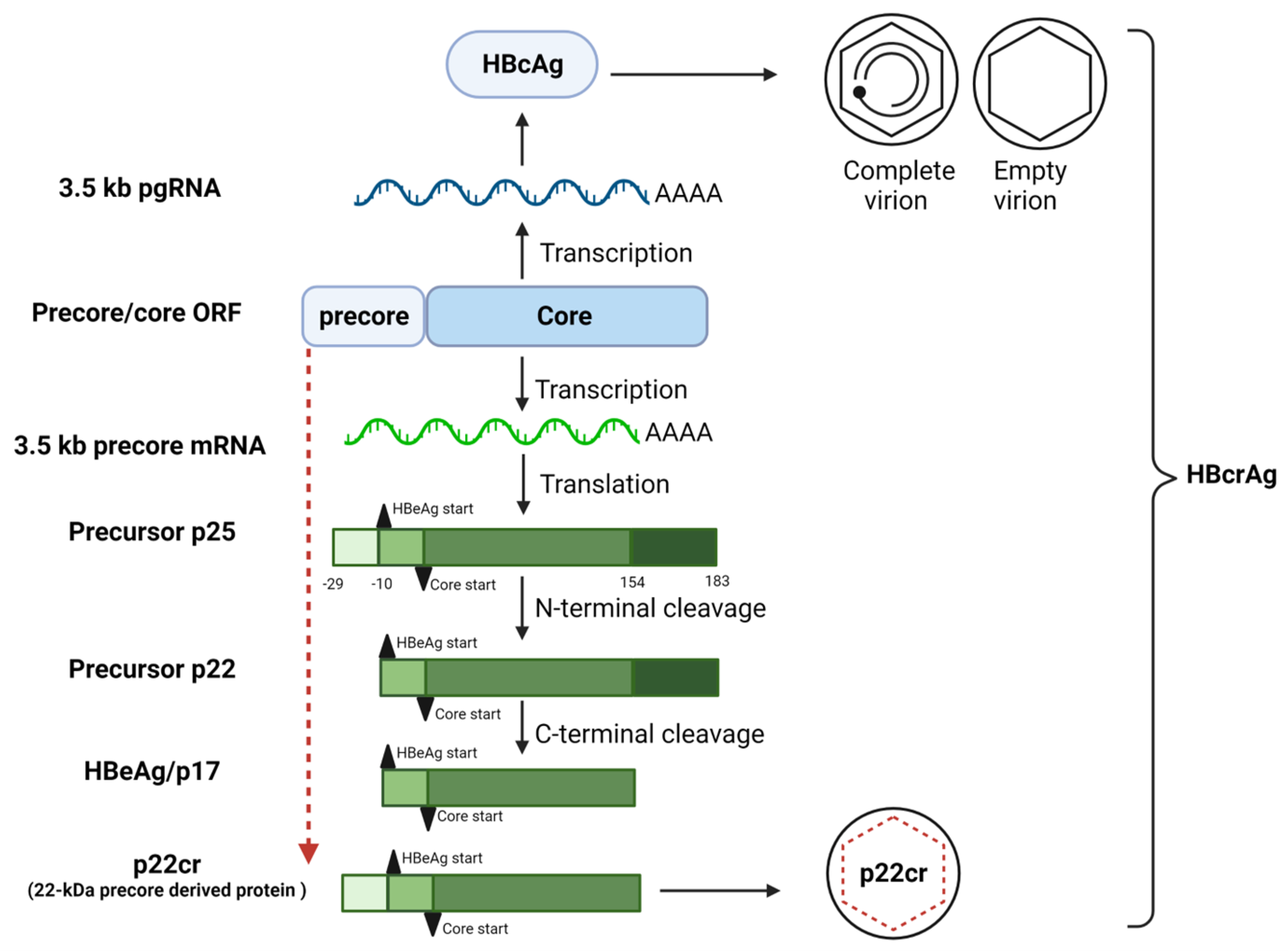
2. Biogenesis of HBeAg
3. Effect of BCP/Precore Mutations on the Expression of HBeAg
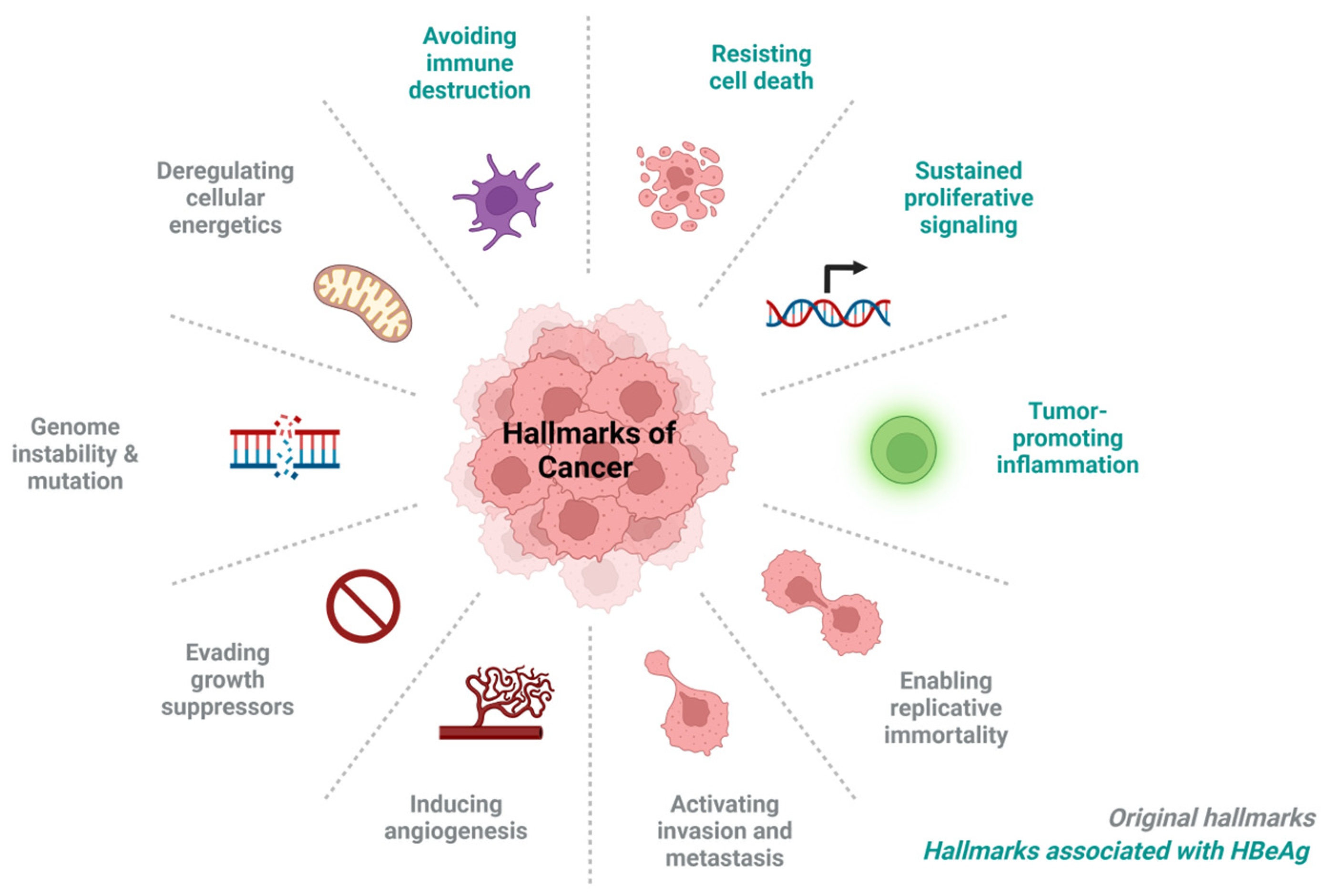
4. HBeAg-Associated Hallmarks of Cancer
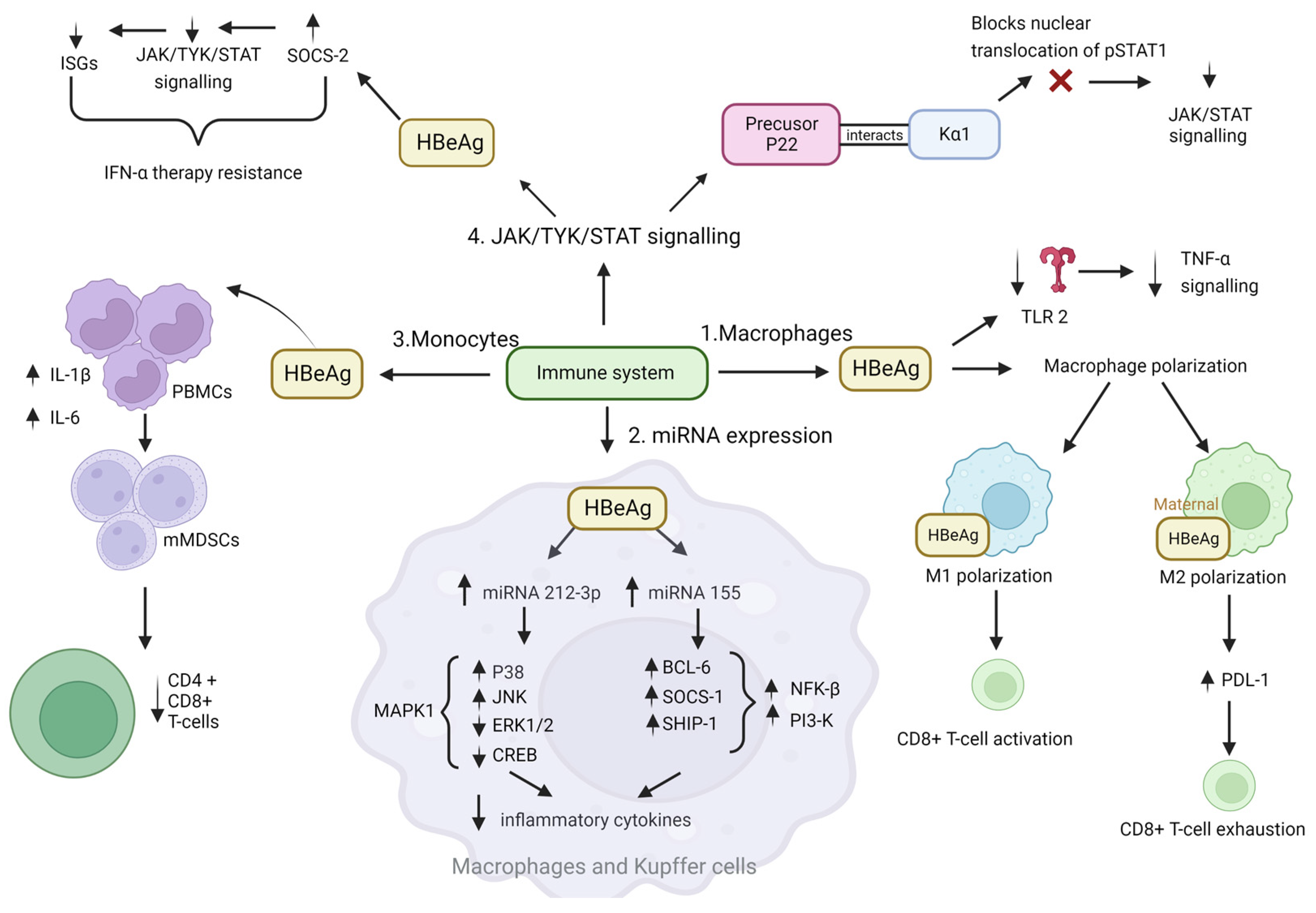
4.1. Immune Evasion, Leading to Persistence
JAK-STAT Pathway
- Mitra and colleagues hypothesised that the reduced antiviral activity of IFN-α against HBV could be due to blockade of the IFN-elicited JAK-STAT pathway by a HBV protein [111]. In their study HBeAg-positive patients exhibited weaker induction of interferon stimulated genes (ISGs) in their livers than HBeAg-negative patients upon IFN-α therapy. Furthermore, the cytosolic HBeAg precursor p22 protein significantly reduced interferon-stimulated response element (ISRE) activity and the expression of ISGs upon IFN-α stimulation in cell culture [111]. p22 did not alter the total STAT1 or pSTAT1 levels in cells treated with IFN-α, but instead blocked the nuclear translocation of pSTAT1 by interacting with karyopherin α1 (Kα1) through its CTD domain, thus impeding JAK-STAT signalling, resulting in host innate immune response evasion and causing resistance to IFN therapy [111].
- Yu and colleagues hypothesised that HBeAg reduced IFN effectiveness and enhanced HBV infection by hijacking the IFN/JAK/STAT pathway [112]. Members of the intracellular suppressor of cytokine signalling (SOCS) family are regulators of cytokine signalling pathways [113,114] that are induced by cytokines and act in a classic negative-feedback loop to inhibit cytokine signalling [115]. SOCS1 and SOCS3 inhibit interferon-mediated antiviral and antiproliferative activities [116,117]. In their study, Yu and colleagues revealed that HBeAg initially activates SOCS2 through the ERK pathway. HBeAg-activated SOCS2 subsequently reduces tyrosine kinase 2 (TYK2) stability, down-regulates IFN receptor expression, represses STAT1 phosphorylation, and finally attenuates ISGs production [112]. Thus, revealing a novel mechanism by which HBeAg is coordinated to enhance HBV replication by hijacking the IFN/JAK/STAT pathway and antiviral action.

4.2. Tumour Promoting Inflammation
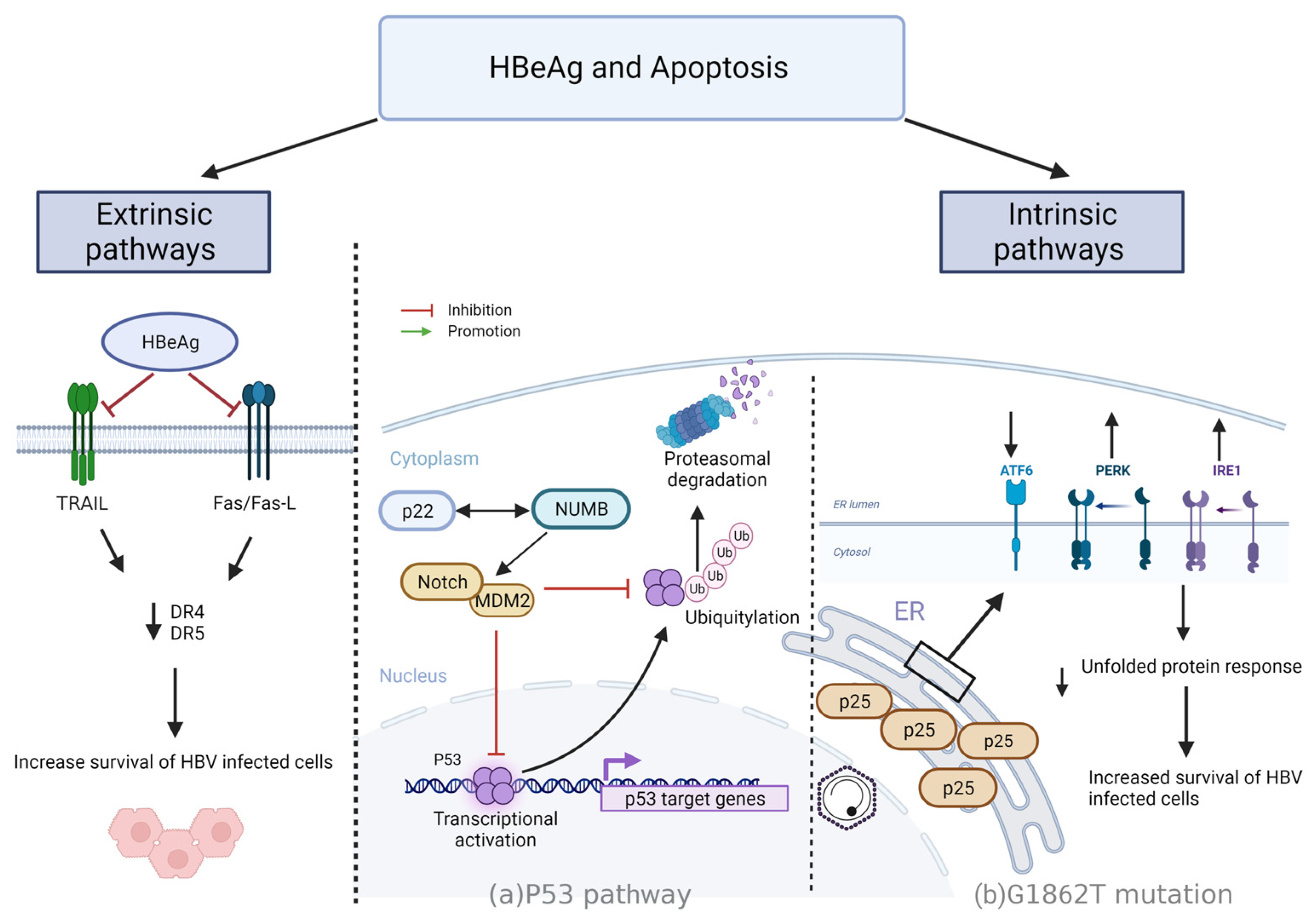
4.3. Resisting Cell Death
4.3.1. Intrinsic Mechanisms
- p53 driven apoptosis
- p25 regulation of apoptosis via UPR
4.3.2. Extrinsic Pathways
- HBeAg regulation of apoptosis via Fas/FasL

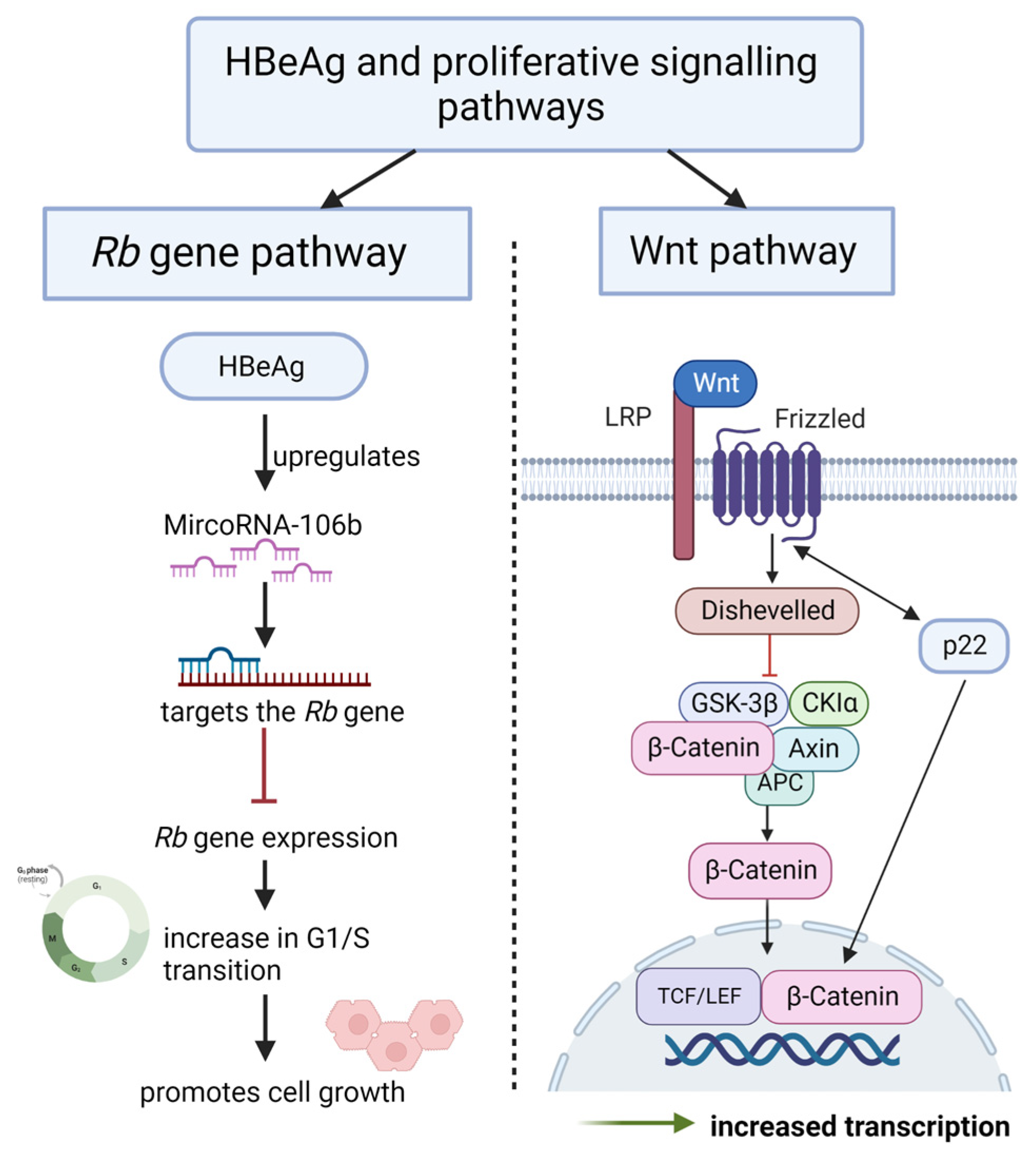
4.4. Promote Sustained Proliferative Signalling
4.4.1. HBeAg Interferes with Wnt/β-catenin Signalling
4.4.2. HBeAg Interferes with the Regulation of the Cell Cycle

5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marion, P.L.; Cullen, J.M.; Azcárraga, R.R.; Van Davelaar, M.J.; Robinson, W.S.; Wai, C.T. Experimental transmission of duck hepatitis B virus to pekin ducks and to domestic geese. Hepatology 1987, 7, 724–731. [Google Scholar] [CrossRef] [PubMed]
- WHO. Hepatitis B Vaccine Epidemology Record; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, T. Multistep carcinogenesis: A 1992 perspective. Science 1992, 258, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Guerrieri, F.; Belloni, L.; Pediconi, N.; Levrero, M.; LaFramboise, T.; Earnest-Silveira, L. Molecular mechanisms of HBV-associated hepatocarcinogenesis. Semin. Liver Dis. 2013, 33, 147–156. [Google Scholar] [CrossRef]
- Jiang, Y.; Han, Q.; Zhao, H.; Zhang, J. The Mechanisms of HBV-Induced Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 435–450. [Google Scholar] [CrossRef]
- Milich, D.R. Is the function of the HBeAg really unknown? Hum. Vaccin. Immunother. 2019, 15, 2187–2191. [Google Scholar] [CrossRef]
- Hossain, M.G.; Akter, S.; Ohsaki, E.; Ueda, K. Impact of the Interaction of Hepatitis B Virus with Mitochondria and Associated Proteins. Viruses 2020, 12, 175. [Google Scholar] [CrossRef]
- Kim, S.-J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B Virus Disrupts Mitochondrial Dynamics: Induces Fission and Mitophagy to Attenuate Apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Torresi, J.; Tran, B.M.; Christiansen, D.; Earnest-Silveira, L.; Schwab, R.H.M.; Vincan, E. HBV-related hepatocarcinogenesis: The role of signalling pathways and innovative ex vivo research models. BMC Cancer 2019, 19, 707. [Google Scholar] [CrossRef]
- Kramvis, A.; Kew, M.C. Epidemiology of hepatitis B virus in Africa, its genotypes and clinical associations of genotypes. Hepatol. Res. 2007, 37, S9–S19. [Google Scholar] [CrossRef]
- Robaczewska, M.; Narayan, R.; Seigneres, B.; Schorr, O.; Thermet, A.; Podhajska, A.J.; Trepo, C.; Zoulim, F.; Nielsen, P.E.; Cova, L. Sequence-specific inhibition of duck hepatitis B virus reverse transcription by peptide nucleic acids (PNA). J. Hepatol. 2005, 42, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Spearman, C.W.; Sonderup, M.W. Preventing hepatitis B and hepatocellular carcinoma in South Africa: The case for a birth-dose vaccine. S. Afr. Med. J. 2014, 104, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Tiollais, P.; Pourcel, C.; Dejean, A. The hepatitis B virus. Nature 1985, 317, 489–495. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef]
- Zhao, F.; Xie, X.; Tan, X.; Yu, H.; Tian, M.; Lv, H.; Qin, C.; Qi, J.; Zhu, Q. The Functions of Hepatitis B Virus Encoding Proteins: Viral Persistence and Liver Pathogenesis. Front. Immunol. 2021, 12, 691766. [Google Scholar] [CrossRef]
- Arbuthnot, P.; Kew, M. Hepatitis B virus and hepatocellular carcinoma. Int. J. Exp. Pathol. 2001, 82, 77–100. [Google Scholar] [CrossRef]
- Sartorius, K.; An, P.; Winkler, C.; Chuturgoon, A.; Li, X.; Makarova, J.; Kramvis, A. The Epigenetic Modulation of Cancer and Immune Pathways in Hepatitis B Virus-Associated Hepatocellular Carcinoma: The Influence of HBx and miRNA Dysregulation. Front. Immunol. 2021, 12, 661204. [Google Scholar] [CrossRef]
- Liu, H.; Xu, L.; He, H.; Zhu, Y.; Liu, J.; Wang, S.; Chen, L.; Wu, Q.; Xu, J.; Gu, J. Hepatitis B virus X protein promotes hepatoma cell invasion and metastasis by stabilizing S nail protein. J. Cancer Sci. 2012, 103, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Lee, Y.M.; Bae, S.K.; Murakami, S.; Yun, Y.; Kim, K.W. Human hepatitis B virus X protein is a possible mediator of hypoxia-induced angiogenesis in hepatocarcinogenesis. Biochem. Biophys. Res. Commun. 2000, 268, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Hung, C.H.; Lee, C.M.; Hu, T.H.; Wang, J.H.; Wang, J.C.; Lu, S.N.; Changchien, C.S. Pre-S Deletion and Complex Mutations of Hepatitis B Virus Related to Advanced Liver Disease in HBeAg-Negative Patients. Gastroenterology 2007, 133, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Mak, D.; Kramvis, A. Molecular characterization of hepatitis B virus isolated from Black South African cancer patients, with and without hepatocellular carcinoma. Arch. Virol. 2020, 165, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Milich, D.; Liang, T.J. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology 2003, 38, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- You, S.L.; Yang, H.I.; Chen, C.J. Seropositivity of hepatitis B e antigen and hepatocellular carcinoma. Ann. Med. 2004, 36, 215–224. [Google Scholar] [CrossRef]
- Kramvis, A.; Chang, K.M.; Dandri, M.; Farci, P.; Glebe, D.; Hu, J.; Janssen, H.L.A.; Lau, D.T.Y.; Penicaud, C.; Pollicino, T.; et al. A roadmap for serum biomarkers for hepatitis B virus: Current status and future outlook. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 727–745. [Google Scholar] [CrossRef]
- Chen, C.Y.; Crowther, C.; Kew, M.C.; Kramvis, A. A valine to phenylalanine mutation in the precore region of hepatitis B virus causes intracellular retention and impaired secretion of HBe-antigen. Hepatol. Res. 2008, 38, 580–592. [Google Scholar] [CrossRef]
- Chen, M.T.; Billaud, J.-N.; Sällberg, M.; Guidotti, L.G.; Chisari, F.V.; Jones, J.; Hughes, J.; Milich, D.R. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc. Natl. Acad. Sci. USA 2004, 101, 14913–14918. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Inverso, D.; Sironi, L.; Di Lucia, P.; Fioravanti, J.; Ganzer, L.; Fiocchi, A.; Vacca, M.; Aiolfi, R.; Sammicheli, S.; et al. Immunosurveillance of the Liver by Intravascular Effector CD8+ T Cells. Cell 2015, 161, 486–500. [Google Scholar] [CrossRef]
- Kramvis, A.; Kew, M.; François, G. Hepatitis B virus genotypes. Vaccine 2005, 23, 2409–2423. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Mertz, J.E. Differential regulation of the pre-C and pregenomic promoters of human hepatitis B virus by members of the nuclear receptor superfamily. J. Virol. 1997, 71, 9366–9374. [Google Scholar] [CrossRef] [PubMed]
- Yuh, C.; Chang, Y.; Ting, L. Transcriptional regulation of precore and pregenomic RNAs of hepatitis B virus. J. Virol. 1992, 66, 4073–4084. [Google Scholar] [CrossRef] [PubMed]
- Guerrieri, F.; Belloni, L.; D’Andrea, D.; Pediconi, N.; Le Pera, L.; Testoni, B.; Scisciani, C.; Floriot, O.; Zoulim, F.; Tramontano, A.; et al. Genome-wide identification of direct HBx genomic targets. BMC Genom. 2017, 18, 184. [Google Scholar] [CrossRef]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- İnan, N.; Tabak, F. Hepatitis B Virus: Biology and Life Cycle. Viral Hepatit Derg. 2015, 21, 1–7. [Google Scholar] [CrossRef]
- Trépo, C.; Chan, H.L.Y.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.-H.; Yeh, C.-T.; Yen, T. Transport of hepatitis B virus precore protein into the nucleus after cleavage of its signal peptide. J. Virol. 1989, 63, 5238–5243. [Google Scholar] [CrossRef]
- Messageot, F.; Salhi, S.; Eon, P.; Rossignol, J.-M. Proteolytic processing of the hepatitis B virus e antigen precursor: Cleavage at two furin consensus sequences. J. Biol. Chem. 2003, 278, 891–895. [Google Scholar] [CrossRef]
- Salhi, S.; Messageot, F.; Carlier, D.; Jean-Jean, O.; Rossignol, J.M. Identification of a cellular protein specifically interacting with the precursor of the hepatitis B e antigen. J. Viral Hepat. 2001, 8, 169–173. [Google Scholar] [CrossRef]
- Yeh, C.-T.; Liaw, Y.; Ou, J.-H. The arginine-rich domain of hepatitis B virus precore and core proteins contains a signal for nuclear transport. J. Virol. 1990, 64, 6141–6147. [Google Scholar] [CrossRef]
- Deroubaix, A.; Kramvis, A. In vitro expression of precore proteins of hepatitis B virus subgenotype A1 is affected by HBcAg, and can affect HBsAg secretion. Sci. Rep. 2021, 11, 8167. [Google Scholar] [CrossRef]
- Alexander, C.G.; Jurgens, M.C.; Shepherd, D.A.; Freund, S.M.; Ashcroft, A.E.; Ferguson, N. Thermodynamic origins of protein folding, allostery, and capsid formation in the human hepatitis B virus core protein. Proc. Natl. Acad. Sci. USA 2013, 110, E2782–E2791. [Google Scholar] [CrossRef]
- Ito, K.; Kim, K.-H.; Lok, A.S.-F.; Tong, S. Characterization of genotype-specific carboxyl-terminal cleavage sites of hepatitis B virus e antigen precursor and identification of furin as the candidate enzyme. J. Virol. 2009, 83, 3507–3517. [Google Scholar] [CrossRef]
- Packianathan, C.; Katen, S.P.; Dann, C.E., 3rd; Zlotnick, A.; Tabak, F.; Tabak, F. Conformational changes in the hepatitis B virus core protein are consistent with a role for allostery in virus assembly. J. Virol. 2010, 84, 1607–1615. [Google Scholar] [CrossRef]
- Venkatakrishnan, B.; Zlotnick, A. The Structural Biology of Hepatitis B Virus: Form and Function. Annu. Rev. Virol. 2016, 3, 429–451. [Google Scholar] [CrossRef]
- Di Bisceglie, A.M. Hepatitis B and hepatocellular carcinoma. Hepatology 2009, 49, S56–S60. [Google Scholar] [CrossRef]
- Magnius, L.O.; Espmark, A. A new antigen complex co-occurring with Australia antigen. Acta Pathol. Microbiol. Scand. 1972, 80, 335–337. [Google Scholar] [CrossRef]
- Hong, X.; Luckenbaugh, L.; Mendenhall, M.; Walsh, R.; Cabuang, L.; Soppe, S.; Revill, P.; Burdette, D.; Feierbach, B.; Delaney, W.; et al. Characterization of Hepatitis B Precore/Core-Related Antigens. J. Virol. 2020, 95, e01594-20. [Google Scholar] [CrossRef]
- Honda, M.; Shirasaki, T.; Terashima, T.; Kawaguchi, K.; Nakamura, M.; Oishi, N.; Wang, X.; Shimakami, T.; Okada, H.; Arai, K.; et al. Hepatitis B Virus (HBV) Core-Related Antigen During Nucleos(t)ide Analog Therapy Is Related to Intra-hepatic HBV Replication and Development of Hepatocellular Carcinoma. J. Infect. Dis. 2016, 213, 1096–1106. [Google Scholar] [CrossRef]
- Mak, L.-Y.; Wong, D.K.-H.; Cheung, K.-S.; Seto, W.-K.; Lai, C.-L.; Yuen, M.-F. Review article: Hepatitis B core-related antigen (HBcrAg): An emerging marker for chronic hepatitis B virus infection. Aliment. Pharmacol. Ther. 2018, 47, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A. Molecular characterisation of the genotypes and mutants of hepatitis B virus from South Africa. S. Afr. J. Epidemiol. Infect. 2008, 23, 29–31. [Google Scholar] [CrossRef]
- Revill, P.; Yuen, L.; Walsh, R.; Perrault, M.; Locarnini, S.; Kramvis, A. Bioinformatic analysis of the hepadnavirus e-antigen and its precursor identifies remarkable sequence conservation in all orthohepadnaviruses. J. Med. Virol. 2010, 82, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A.; Arakawa, K.; Yu, M.C.; Nogueira, R.; Stram, D.O.; Kew, M.C. Relationship of serological subtype, basic core promoter and precore mutations to genotypes/subgenotypes of hepatitis B virus. J. Med. Virol. 2008, 80, 27–46. [Google Scholar] [CrossRef]
- Carman, W.; Hadziyannis, S.; McGarvey, M.; Jacyna, M.; Karayiannis, P.; Makris, A.; Thomas, H. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet 1989, 334, 588–591. [Google Scholar] [CrossRef]
- Rezende, R.E.; Fonseca, B.A.; Ramalho, L.N.; Zucoloto, S.; Pinho, J.R.R.; Bertolini, D.A.; Martinelli, A.L. The precore mutation is associated with severity of liver damage in Brazilian patients with chronic hepatitis B. J. Clin. Virol. 2005, 32, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Xie, J.; Liu, S.; Zhang, H.; Han, L.; Lu, W.; Shen, Q.; Xu, G.; Dong, H.; Shen, J. Association between the various mutations in viral core promoter region to different stages of hepatitis B, ranging of asymptomatic carrier state to hepatocellular carcinoma. Off. J. Am. Coll. Gastroenterol. 2011, 106, 81–92. [Google Scholar] [CrossRef]
- Revill, P.A.; Locarnini, S.A. New perspectives on the hepatitis B virus life cycle in the human liver. J. Clin. Investig. 2016, 126, 833–836. [Google Scholar] [CrossRef]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef]
- Baptista, M.; Kramvis, A.; Kew, M.C. High prevalence of 1762T 1764A mutations in the basic core promoter of hepatitis B virus isolated from black Africans with hepatocellular carcinoma compared with asymptomatic carriers. Hepatology 1999, 29, 946–953. [Google Scholar] [CrossRef]
- Buckwold, V.E.; Xu, Z.; Chen, M.; Yen, T.; Ou, J.-H. Effects of a naturally occurring mutation in the hepatitis B virus basal core promoter on precore gene expression and viral replication. J. Virol. 1996, 70, 5845–5851. [Google Scholar] [CrossRef] [PubMed]
- Bell, T.G.; Yousif, M.; Kramvis, A. Bioinformatic curation and alignment of genotyped hepatitis B virus (HBV) sequence data from the GenBank public database. Springerplus 2016, 5, 1896. [Google Scholar] [CrossRef] [PubMed]
- Orito, E.; Ichida, T.; Sakugawa, H.; Sata, M.; Horiike, N.; Hino, K.; Okita, K.; Okanoue, T.; Iino, S.; Tanaka, E.; et al. Geographic distribution of hepatitis B virus (HBV) genotype in patients with chronic HBV infection in Japan. Hepatology 2001, 34, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Hasegawa, I.; Kato, T.; Orito, E.; Hirashima, N.; Acharya, S.K.; Gish, R.G.; Kramvis, A.; Kew, M.C.; Yoshihara, N.; et al. A case-control study for differences among hepatitis B virus infections of genotypes A (subtypes Aa and Ae) and D. J. Hepatol. 2004, 40, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.; Biswas, A.; Datta, S.; Banerjee, A.; Panigrahi, R.; Chakrabarti, S.; De, B.; Chakravarty, R. Subgenotypes of hepatitis B virus genotype D (D1, D2, D3 and D5) in India: Differential pattern of mutations, liver injury and occult HBV infection. J. Viral Hepat. 2009, 16, 749–756. [Google Scholar] [CrossRef]
- Schaefer, S. Hepatitis B virus genotypes in Europe. Hepatol. Res. 2007, 37, S20–S26. [Google Scholar] [CrossRef]
- Kobayashi, M.; Suzuki, F.; Arase, Y.; Akuta, N.; Suzuki, Y.; Hosaka, T.; Saitoh, S.; Kobayashi, M.; Tsubota, A.; Someya, T. Infection with hepatitis B virus genotype A in Tokyo, Japan during 1976 through 2001. J. Gastroenterol. 2004, 39, 844–850. [Google Scholar] [CrossRef]
- Wai, C.T.; Mak, B.; Chua, W.; Tan, M.H.; Ng, S.; Cheok, A.; Wong, M.L.; Lim, S.G. Misperceptions among patients with chronic hepatitis B in Singapore. World J. Gastroenterol. 2005, 11, 5002–5005. [Google Scholar] [CrossRef]
- McMahon, B.J. Epidemiology and natural history of hepatitis B. Semin. Liver Dis. 2005, 25, 3–8. [Google Scholar] [CrossRef]
- Kramvis, A. Molecular characteristics and clinical relevance of African genotypes and subgenotypes of hepatitis B virus. S. Afr. Med. J. 2018, 108, 17–21. [Google Scholar] [CrossRef]
- Parekh, S.; Zoulim, F.; Ahn, S.H.; Tsai, A.; Li, J.; Kawai, S.; Khan, N.; Trépo, C.; Wands, J.; Tong, S. Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J. Virol. 2003, 77, 6601–6612. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Kramvis, A.; Kawai, S.; Spangenberg, H.C.; Li, J.; Kimbi, G.; Kew, M.; Wands, J.; Tong, S.J.G. Sequence variation upstream of precore translation initiation codon reduces hepatitis B virus e antigen production. Gastroenterology 2003, 125, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Bhoola, N.H.; Kramvis, A. Hepatitis B e Antigen Expression by Hepatitis B Virus Subgenotype A1 Relative to Subgenotypes A2 and D3 in Cultured Hepatocellular Carcinoma (Huh7) Cells. Intervirology 2016, 59, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.; Engelbrecht, J.; Brunak, S.; von Heijne, G.; Pa, M.; Earnest-Silveira, L. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997, 10, 673–676. [Google Scholar] [CrossRef]
- Mitra, B. Biological Functions of Intracellular Hepatitis B e Antigen; Indiana University-Purdue University Indianapolis: Indianapolis, IN, USA, 2019. [Google Scholar]
- Malik, A.; Kumar, D.; Khan, A.A.; Khan, A.A.; Chaudhary, A.A.; Husain, S.A.; Kar, P. Hepatitis B virus precore G1896A mutation in chronic liver disease patients with HBeAg negative serology from North India. Saudi J. Biol. Sci. 2018, 25, 1257–1262. [Google Scholar] [CrossRef]
- Kramvis, A.; Kostaki, E.-G.; Hatzakis, A.; Paraskevis, D. Immunomodulatory Function of HBeAg Related to Short-Sighted Evolution, Transmissibility, and Clinical Manifestation of Hepatitis B Virus. Front. Microbiol. 2018, 9, 2521. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Tian, Y.; Kuo, C.-f.; Akbari, O.; Ou, J.H.J. Maternal-Derived Hepatitis B Virus e Antigen Alters Macrophage Function in Offspring to Drive Viral Persistence after Vertical Transmission. Immunity 2016, 44, 1204–1214. [Google Scholar] [CrossRef]
- Wang, J.; Yu, Y.; Li, G.; Shen, C.; Meng, Z.; Zheng, J.; Jia, Y.; Chen, S.; Zhang, X.; Zhu, M.; et al. Relationship between serum HBV-RNA levels and intrahepatic viral as well as histologic activity markers in entecavir-treated patients. J. Hepatol. 2018, 68, 16–24. [Google Scholar] [CrossRef]
- Ferrando-Martinez, S.; Huang, K.; Bennett, A.S.; Sterba, P.; Yu, L.; Suzich, J.A.; Janssen, H.L.A.; Robbins, S.H. HBeAg seroconversion is associated with a more effective PD-L1 blockade during chronic hepatitis B infection. JHEP Rep. 2019, 1, 170–178. [Google Scholar] [CrossRef]
- Liaw, Y.-F.; Lau, G.K.K.; Kao, J.-H.; Gane, E. Hepatitis B e Antigen Seroconversion: A Critical Event in Chronic Hepatitis B Virus Infection. Dig. Dis. Sci. 2010, 55, 2727–2734. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Huang, R.; Chen, Y.; Liu, Y.; Wang, J.; Yan, X.; Zhang, Z.; Wu, C. Profiles of serum soluble programmed death-1 and programmed death-ligand 1 levels in chronic hepatitis B virus-infected patients with different disease phases and after anti-viral treatment. Aliment. Pharmacol. Ther. 2020, 51, 1180–1187. [Google Scholar] [CrossRef]
- Chen, W.; Bian, H.; Xie, X.; Yang, X.; Bi, B.; Li, C.; Zhang, Y.; Zhu, Q.; Song, J.; Qin, C.; et al. Negative feedback loop of ERK/CREB/miR-212-3p inhibits HBeAg-induced macrophage activation. J. Cell. Mol. Med. 2020, 24, 10935–10945. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, X.; Wang, J. MicroRNA-212 Targets Mitogen-Activated Protein Kinase 1 to Inhibit Proliferation and Invasion of Prostate Cancer Cells. Oncol. Res. 2018, 26, 1093–1102. [Google Scholar] [CrossRef]
- Wang, W.; Bian, H.; Li, F.; Li, X.; Zhang, D.; Sun, S.; Song, S.; Zhu, Q.; Ren, W.; Qin, C.; et al. HBeAg induces the expression of macrophage miR-155 to accelerate liver injury via promoting production of inflammatory cytokines. Cell. Mol. Life Sci. 2018, 75, 2627–2641. [Google Scholar] [CrossRef] [PubMed]
- Subedi, A.; Park, P.-H. Autocrine and paracrine modulation of microRNA-155 expression by globular adiponectin in RAW 264.7 macrophages: Involvement of MAPK/NF-κB pathway. Cytokine 2013, 64, 638–641. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Alivernini, S.; Ballantine, L.E.; Asquith, D.L.; Millar, N.L.; Gilchrist, D.S.; Reilly, J.; Ierna, M.; Fraser, A.R.; Stolarski, B.; et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 11193–11198. [Google Scholar] [CrossRef]
- Park, E.J.; Shen, L.; Sun, D.; Pezzuto, J.M. Inhibitory effect of a callophycin A derivative on iNOS expression via inhibition of Akt in lipopolysaccharide-stimulated RAW 264.7 cells. J. Nat. Prod. 2014, 77, 527–535. [Google Scholar] [CrossRef]
- Du, F.; Yu, F.; Wang, Y.; Hui, Y.; Carnevale, K.; Fu, M.; Lu, H.; Fan, D. MicroRNA-155 deficiency results in decreased macrophage inflammation and attenuated atherogenesis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 759–767. [Google Scholar] [CrossRef]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J. Clin. Investig. 2012, 122, 4190–4202. [Google Scholar] [CrossRef]
- Xie, X.; Lv, H.; Liu, C.; Su, X.; Yu, Z.; Song, S.; Bian, H.; Tian, M.; Qin, C.; Qi, J.; et al. HBeAg mediates inflammatory functions of macrophages by TLR2 contributing to hepatic fibrosis. BMC Med. 2021, 19, 247. [Google Scholar] [CrossRef]
- Jegaskanda, S.; Ahn, S.H.; Skinner, N.; Thompson, A.J.; Ngyuen, T.; Holmes, J.; De Rose, R.; Navis, M.; Winnall, W.R.; Kramski, M.; et al. Downregulation of interleukin-18-mediated cell signaling and interferon gamma expression by the hepatitis B virus e antigen. J. Virol. 2014, 88, 10412–10420. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, L.; Yang, G.; Zhan, J.; Guo, L.; Chen, Y.; Fan, C.; Liu, D.; Guo, D. Hepatitis B e Antigen Inhibits NF-κB Activity by Interrupting K63-Linked Ubiquitination of NEMO. J. Virol. 2019, 93, e00667-18. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; La Motte-Mohs, R.N.A.; Rudolph, D.; Zúñiga-Pflücker, J.C.; Mak, T.W. The role of nuclear factor-κB essential modulator (NEMO) in B cell development and survival. Proc. Natl. Acad. Sci. USA 2003, 100, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, A.; Karayiannis, P. HBeAg negative variants and their role in the natural history of chronic hepatitis B virus infection. World J. Gastroenterol. 2014, 20, 7644–7652. [Google Scholar] [CrossRef] [PubMed]
- Leu, C.-M.; Lu, Y.-C.; Peng, W.-L.; Chu, H.-T.; Hu, C.-P. The hepatitis B virus e antigen suppresses the respiratory burst and mobility of human monocytes and neutrophils. Immunobiology 2014, 219, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Zilio, S.; Marigo, I.; Dolcetti, L.; Zanovello, P.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr. Opin. Immunol. 2010, 22, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yu, X.; Zhou, C.; Mao, R.; Zhu, M.; Zhu, H.; Ma, Z.; Mitra, B.; Zhao, G.; Huang, Y.; et al. Hepatitis B e antigen induces the expansion of monocytic myeloid-derived suppressor cells to dampen T-cell function in chronic hepatitis B virus infection. PLoS Pathog. 2019, 15, e1007690. [Google Scholar] [CrossRef]
- Martinet, J.; Dufeu–Duchesne, T.; Bruder Costa, J.; Larrat, S.; Marlu, A.; Leroy, V.; Plumas, J.; Aspord, C. Altered Functions of Plasmacytoid Dendritic Cells and Reduced Cytolytic Activity of Natural Killer Cells in Patients With Chronic HBV Infection. Gastroenterology 2012, 143, 1586–1596. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Riordan, S.M.; Skinner, N.; Kurtovic, J.; Locarnini, S.; Visvanathan, K.J.C.; Immunology, V. Reduced expression of toll-like receptor 2 on peripheral monocytes in patients with chronic hepatitis B. Clin. Vaccine Immunol. 2006, 13, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, K.; Skinner, N.A.; Thompson, A.J.V.; Riordan, S.M.; Sozzi, V.; Edwards, R.; Rodgers, S.; Kurtovic, J.; Chang, J.; Lewin, S.; et al. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 2007, 45, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, K.; Dusheiko, G.; Giles, M.; Wong, M.-L.; Phung, N.; Walker, S.; Le, S.; Lim, S.G.; Gane, E.; Ngu, M.; et al. Managing HBV in pregnancy. Prevention, prophylaxis, treatment and follow-up: Position paper produced by Australian, UK and New Zealand key opinion leaders. Gut 2016, 65, 340. [Google Scholar] [CrossRef]
- Lang, T.; Lo, C.; Skinner, N.; Locarnini, S.; Visvanathan, K.; Mansell, A. The Hepatitis B e antigen (HBeAg) targets and suppresses activation of the Toll-like receptor signaling pathway. J. Hepatol. 2011, 55, 762–769. [Google Scholar] [CrossRef]
- Milich, D.R.; Chen, M.K.; Hughes, J.L.; Jones, J.E. The Secreted Hepatitis B Precore Antigen Can Modulate the Immune Response to the Nucleocapsid: A Mechanism for Persistence. J. Immunol. 1998, 160, 2013. [Google Scholar] [CrossRef]
- Janssen, H.L.A.; van Zonneveld, M.; Senturk, H.; Zeuzem, S.; Akarca, U.S.; Cakaloglu, Y.; Simon, C.; So, T.M.K.; Gerken, G.; de Man, R.A.; et al. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: A randomised trial. Lancet 2005, 365, 123–129. [Google Scholar] [CrossRef]
- Aikawa, T.; Kanai, K.; Kako, M.; Kawasaki, T.; Hino, K.; Iwabuchi, S.; Tsubouchi, H.; Takehira, Y.; Tsuda, F.; Okamoto, H.; et al. Interferon-α2a for chronic hepatitis B with e antigen or antibody: Comparable antiviral effects on wild-type virus and precore mutant. J. Viral Hepat. 1995, 2, 243–250. [Google Scholar] [CrossRef]
- Heijtink, R.A.; Janssen, H.L.A.; Hop, W.C.J.; Osterhaus, A.D.M.E.; Schalm, S.W. Interferon-alpha therapy for chronic hepatitis B: Early response related to pre-treatment changes in viral replication. J. Med. Virol. 2001, 63, 217–219. [Google Scholar] [CrossRef]
- Mitra, B.; Wang, J.; Kim Elena, S.; Mao, R.; Dong, M.; Liu, Y.; Zhang, J.; Guo, H. Hepatitis B Virus Precore Protein p22 Inhibits Alpha Interferon Signaling by Blocking STAT Nuclear Translocation. J. Virol. 2019, 93, e00196-19. [Google Scholar] [CrossRef]
- Yu, Y.; Wan, P.; Cao, Y.; Zhang, W.; Chen, J.; Tan, L.; Wang, Y.; Sun, Z.; Zhang, Q.; Wan, Y.; et al. Hepatitis B Virus e Antigen Activates the Suppressor of Cytokine Signaling 2 to Repress Interferon Action. Sci. Rep. 2017, 7, 1729. [Google Scholar] [CrossRef]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef]
- Starr, R.; Willson, T.A.; Viney, E.M.; Murray, L.J.L.; Rayner, J.R.; Jenkins, B.J.; Gonda, T.J.; Alexander, W.S.; Metcalf, D.; Nicola, N.A.; et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997, 387, 917–921. [Google Scholar] [CrossRef]
- Piessevaux, J.; Lavens, D.; Peelman, F.; Tavernier, J. The many faces of the SOCS box. Cytokine Growth Factor Rev. 2008, 19, 371–381. [Google Scholar] [CrossRef]
- Song, B.-C.; Suh, D.J.; Lee, H.C.; Chung, Y.-H.; Lee, Y.S. Hepatitis B e Antigen Seroconversion After Lamivudine Therapy Is Not Durable in Patients With Chronic Hepatitis B in Korea. Hepatology 2000, 32, 803–806. [Google Scholar] [CrossRef]
- Collins, A.S.; Ahmed, S.; Napoletano, S.; Schroeder, M.; Johnston, J.A.; Hegarty, J.E.; O’Farrelly, C.; Stevenson, N.J. Hepatitis C virus (HCV)-induced suppressor of cytokine signaling (SOCS) 3 regulates proinflammatory TNF-α responses. J. Leukoc. Biol. 2014, 96, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Kundu, J.K.; Surh, Y.J. Inflammation: Gearing the journey to cancer. Mutat. Res. 2008, 659, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Pollicino, T.; Petersen, J.; Belloni, L.; Raimondo, G.; Dandri, M. Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 2009, 51, 581–592. [Google Scholar] [CrossRef]
- Frelin, L.; Wahlström, T.; Tucker, A.E.; Jones, J.; Hughes, J.; Lee, B.O.; Billaud, J.N.; Peters, C.; Whitacre, D.; Peterson, D.; et al. A mechanism to explain the selection of the hepatitis e antigen-negative mutant during chronic hepatitis B virus infection. J. Virol. 2009, 83, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Bhoola, N.H.; Kramvis, A. Expression of wild-type or G1862T mutant HBe antigen of subgenotype A1 of hepatitis B virus and the unfolded protein response in Huh7 cells. J. Gen. Virol. 2017, 98, 1422–1433. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Apoptosis-targeted therapies for cancer. Cancer Cell 2003, 3, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Tait, S. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Caruso, S.; O’Brien, D.R.; Cleary, S.P.; Roberts, L.R.; Zucman-Rossi, J. Genetics of Hepatocellular Carcinoma: Approaches to Explore Molecular Diversity. Hepatology 2021, 73, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Cui, L.; Wang, Y.; Yang, G.; He, J.; Hao, R.; Fan, C.; Qu, M.; Liu, Z.; Wang, M.; et al. Hepatitis B e antigen and its precursors promote the progress of hepatocellular carcinoma by interacting with NUMB and decreasing p53 activity. Hepatology 2016, 64, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Colaluca, I.N.; Tosoni, D.; Nuciforo, P.; Senic-Matuglia, F.; Galimberti, V.; Viale, G.; Pece, S.; Di Fiore, P.P. NUMB controls p53 tumour suppressor activity. Nature 2008, 451, 76–80. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.A.; McGlade, C.J. Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J. Biol. Chem. 2003, 278, 23196–23203. [Google Scholar] [CrossRef]
- McGill, M.A.; Dho, S.E.; Weinmaster, G.; McGlade, C.J. Numb regulates post-endocytic trafficking and degradation of Notch1. J. Biol. Chem. 2009, 284, 26427–26438. [Google Scholar] [CrossRef]
- Tsai, K.-N.; Ou, J.-H.J. Hepatitis B virus e antigen and viral persistence. Curr. Opin. Virol. 2021, 51, 158–163. [Google Scholar] [CrossRef]
- Hou, J.; Lin, Y.; Waters, J.; Wang, Z.; Min, J.; Liao, H.; Jiang, J.; Chen, J.; Luo, K.; Karayiannis, P. Detection and significance of a G1862T variant of hepatitis B virus in Chinese patients with fulminant hepatitis. J. Gen. Virol. 2002, 83, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Kostova, Z.; Wolf, D.H. For whom the bell tolls: Protein quality control of the endoplasmic reticulum and the ubiquitin–proteasome connection. EMBO J. 2003, 22, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Kramvis, A.; Kew, M.C.; Bukofzer, S. Hepatitis B virus precore mutants in serum and liver of Southern African Blacks with hepatocellular carcinoma. J. Hepatol. 1998, 28, 132–141. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Apoptosis: A mechanism of acute and chronic liver injury. Gut 2005, 54, 1024–1033. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef] [PubMed]
- Jodo, S.; Kobayashi, S.; Nakajima, Y.; Matsunaga, T.; Nakayama, N.; Ogura, N.; Kayagaki, N.; Okumura, K.; Koike, T. Elevated serum levels of soluble Fas/APO-1 (CD95) in patients with hepatocellular carcinoma. Clin. Exp. Immunol. 2001, 112, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Rivero, M.; Crespo, J.; Fábrega, E.; Casafont, F.; Mayorga, M.; Gomez-Fleitas, M.; Pons-Romero, F. Apoptosis mediated by the Fas system in the fulminant hepatitis by hepatitis B virus. J. Viral Hepat. 2002, 9, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef]
- Müller, M.; Strand, S.; Hug, H.; Heinemann, E.M.; Walczak, H.; Hofmann, W.J.; Stremmel, W.; Krammer, P.H.; Galle, P.R. Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. J. Clin. Investig. 1997, 99, 403–413. [Google Scholar] [CrossRef]
- Liu, W.; Guo, T.-F.; Jing, Z.-T.; Tong, Q.-Y. Repression of Death Receptor–Mediated Apoptosis of Hepatocytes by Hepatitis B Virus e Antigen. Am. J. Pathol. 2019, 189, 2181–2195. [Google Scholar] [CrossRef]
- Zaman, S.; Wang, R.; Gandhi, V. Targeting the apoptosis pathway in hematologic malignancies. Leuk. Lymphoma 2014, 55, 1980–1992. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Söderholm, S.; Cantù, C. The WNT/β-catenin dependent transcription: A tissue-specific business. WIREs Mech. Dis. 2021, 13, e1511. [Google Scholar] [CrossRef]
- Flanagan, D.J.; Vincan, E.; Phesse, T.J. Wnt Signaling in Cancer: Not a Binary ON:OFF Switch. Cancer Res. 2019, 79, 5901–5906. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.M.; Flanagan, D.J.; Ebert, G.; Warner, N.; Tran, H.; Fifis, T.; Kastrappis, G.; Christophi, C.; Pellegrini, M.; Torresi, J.; et al. The Hepatitis B Virus Pre-Core Protein p22 Activates Wnt Signaling. Cancers 2020, 12, 1435. [Google Scholar] [CrossRef]
- Damania, P.; Sen, B.; Dar, S.B.; Kumar, S.; Kumari, A.; Gupta, E.; Sarin, S.K.; Venugopal, S.K. Hepatitis B virus induces cell proliferation via HBx-induced microRNA-21 in hepatocellular carcinoma by targeting programmed cell death protein4 (PDCD4) and phosphatase and tensin homologue (PTEN). PLoS ONE 2014, 9, e91745. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.J.; Malumbres, M. MicroRNAs and the cell cycle. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 592–601. [Google Scholar] [CrossRef]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010, 64, 123. [Google Scholar] [CrossRef]
- Winther, T.N.; Jacobsen, K.S.; Mirza, A.H.; Heiberg, I.L.; Bang-Berthelsen, C.H.; Pociot, F.; Hogh, B. Circulating MicroRNAs in Plasma of Hepatitis B e Antigen Positive Children Reveal Liver-Specific Target Genes. Int. J. Hepatol. 2014, 2014, 791045. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Samal, J.; Kandpal, M.; Vivekanandan, P. HBeAg-induced miR-106b promotes cell growth by targeting the retinoblastoma gene. Sci. Rep. 2017, 7, 14371. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Xie, Y. Hepatitis B Virus-Associated Hepatocellular Carcinoma. Adv. Exp. Med. Biol. 2017, 1018, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Durst, M.; Gissmann, M.; Hcenberg, H. A papillomavirus DNA from a carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Nat. Acad. Sci. USA 1983, 80, 3812–3815. [Google Scholar] [CrossRef]
- Vousden, K.H. Regulation of the cell cycle by viral oncoproteins. Semin. Cancer Biol. 1995, 6, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Chisari, F. Viruses, Immunity, and Cancer: Lessons from Hepatitis B. Am. J. Pathol. 2000, 156, 1117–1132. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padarath, K.; Deroubaix, A.; Kramvis, A. The Complex Role of HBeAg and Its Precursors in the Pathway to Hepatocellular Carcinoma. Viruses 2023, 15, 857. https://doi.org/10.3390/v15040857
Padarath K, Deroubaix A, Kramvis A. The Complex Role of HBeAg and Its Precursors in the Pathway to Hepatocellular Carcinoma. Viruses. 2023; 15(4):857. https://doi.org/10.3390/v15040857
Chicago/Turabian StylePadarath, Kiyasha, Aurélie Deroubaix, and Anna Kramvis. 2023. "The Complex Role of HBeAg and Its Precursors in the Pathway to Hepatocellular Carcinoma" Viruses 15, no. 4: 857. https://doi.org/10.3390/v15040857
APA StylePadarath, K., Deroubaix, A., & Kramvis, A. (2023). The Complex Role of HBeAg and Its Precursors in the Pathway to Hepatocellular Carcinoma. Viruses, 15(4), 857. https://doi.org/10.3390/v15040857