

A Novel Dependoparvovirus Identified in Cloacal Swabs of Monk Parakeet (Myiopsitta monachus) from Urban Areas of Spain

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Treatment, DNA Isolation, and Metagenomic Analysis
2.3. Genomic Characterization and Recombination Analysis
2.4. Phylogenetic Analysis and Species Demarcation
2.5. DNA Extraction and Screening of Dependoparvovirus in M. monachus
3. Results
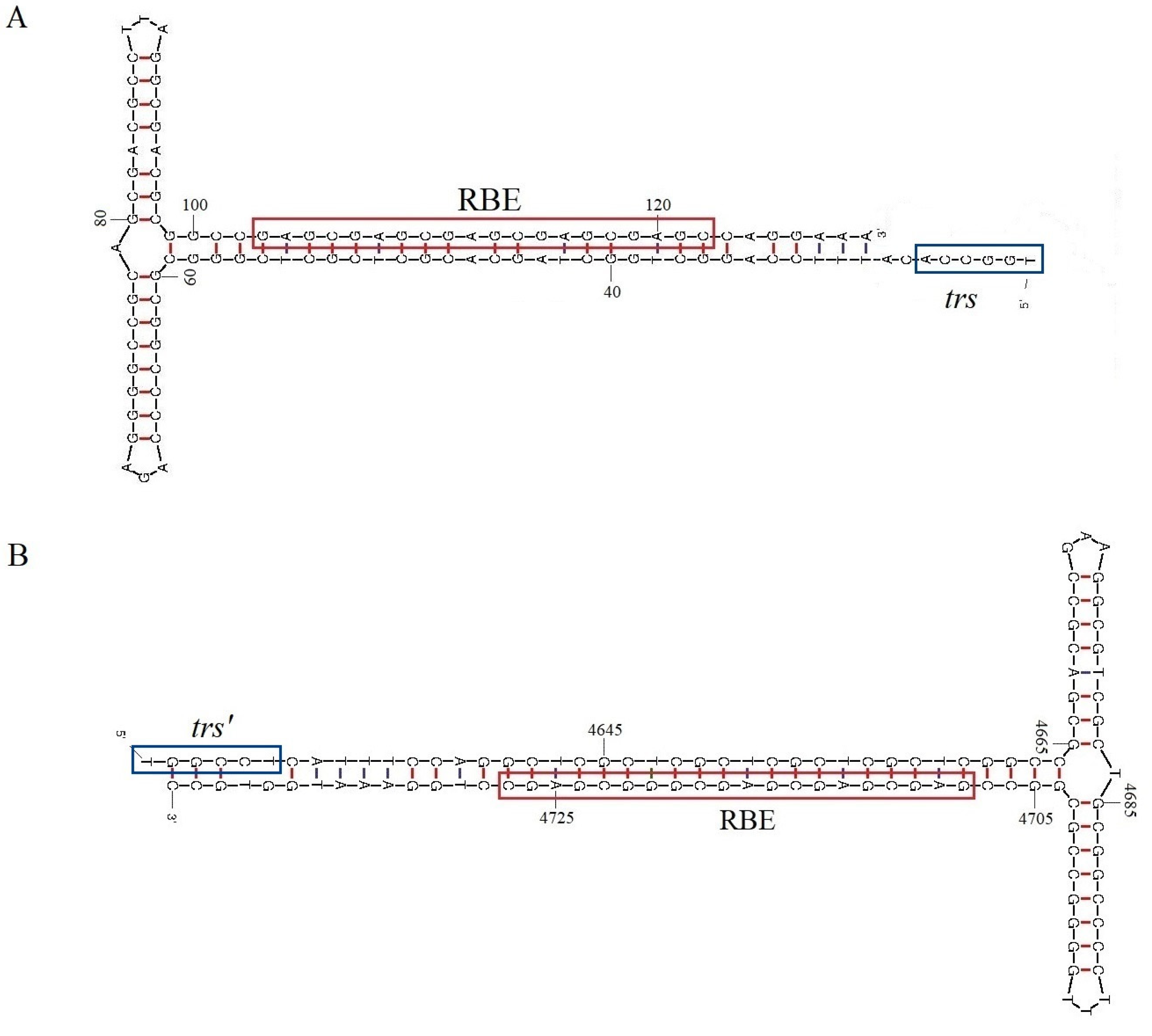
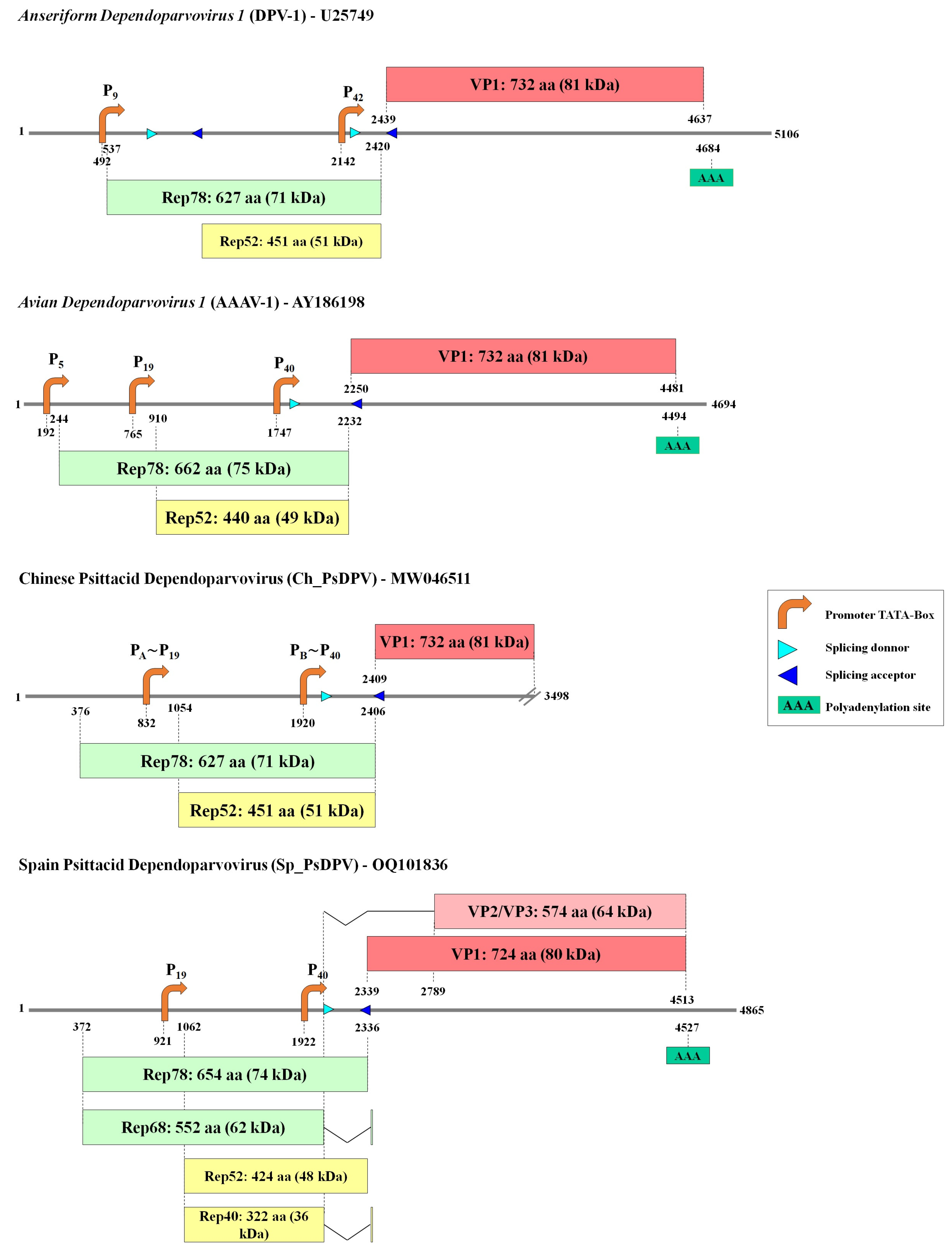
3.1. Sequence, Genomic Organization, and Recombination Analysis
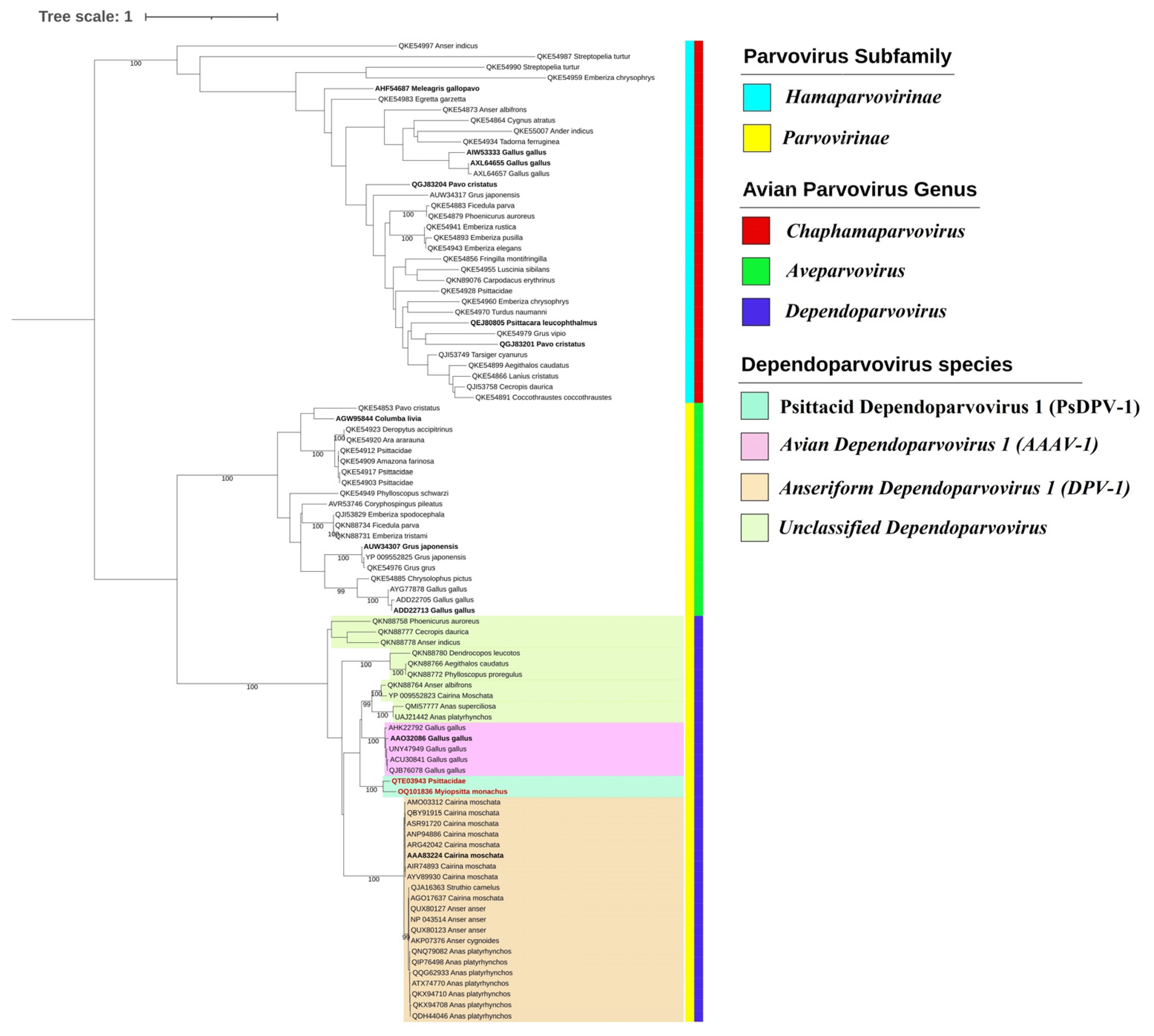
3.2. Phylogenetic and Species Demarcation Analysis
3.3. Screening of Dependoparvovirus in Monk Parakeet
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Diagne, C.; Leroy, B.; Vaissière, A.-C.; Gozlan, R.E.; Roiz, D.; Jarić, I.; Salles, J.-M.; Bradshaw, C.J.A.; Courchamp, F. High and Rising Economic Costs of Biological Invasions Worldwide. Nature 2021, 592, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rohr, J.; Cui, R.; Xin, Y.; Han, L.; Yang, X.; Gu, S.; Du, Y.; Liang, J.; Wang, X.; et al. Biological Invasions Facilitate Zoonotic Disease Emergences. Nat. Commun. 2022, 13, 1762. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; To, K.K.-W.; Chen, H.; Yuen, K.-Y. Cross-Species Transmission and Emergence of Novel Viruses from Birds. Curr. Opin. Virol. 2015, 10, 63–69. [Google Scholar] [CrossRef]
- Vibin, J.; Chamings, A.; Collier, F.; Klaassen, M.; Nelson, T.M.; Alexandersen, S. Metagenomics Detection and Characterisation of Viruses in Faecal Samples from Australian Wild Birds. Sci. Rep. 2018, 8, 8686. [Google Scholar] [CrossRef]
- Briceño, C.; Sandoval-Rodríguez, A.; Yévenes, K.; Larraechea, M.; Morgado, A.; Chappuzeau, C.; Muñoz, V.; Dufflocq, P.; Olivares, F. Interactions between Invasive Monk Parakeets (Myiopsitta monachus) and Other Bird Species during Nesting Seasons in Santiago, Chile. Animals 2019, 9, 923. [Google Scholar] [CrossRef]
- Postigo, J.-L.; Strubbe, D.; Mori, E.; Ancillotto, L.; Carneiro, I.; Latsoudis, P.; Menchetti, M.; Pârâu, L.G.; Parrott, D.; Reino, L.; et al. Mediterranean versus Atlantic Monk Parakeets Myiopsitta monachus: Towards Differentiated Management at the European Scale. Pest Manag. Sci. 2019, 75, 915–922. [Google Scholar] [CrossRef]
- Molina, B.; Postigo, J.L.; Muñoz, A.-R.; del Moral, J.C. La Cotorra Argentina En España, Población Reproductora En 2015 y Método de Censo; SEO/BirdLife: Madrid, Spain, 2016; p. 46. [Google Scholar] [CrossRef]
- Hu, X.; Cai, D.; Liu, S.; Li, Y.; Chen, L.; Luo, G.; Pu, H.; He, Y.; Liu, X.; Zhao, L.; et al. Molecular Characterization of a Novel Budgerigar Fledgling Disease Virus Strain from Budgerigars in China. Front. Vet. Sci. 2022, 8, 813397. [Google Scholar] [CrossRef]
- Blanco, G.; Morinha, F.; Carrete, M.; Tella, J.L. Apparent Lack of Circovirus Transmission from Invasive Parakeets to Native Birds. Int. J. Environ. Res. Public Health 2022, 19, 3196. [Google Scholar] [CrossRef]
- Kessler, S.; Heenemann, K.; Krause, T.; Twietmeyer, S.; Fuchs, J.; Lierz, M.; Corman, V.M.; Vahlenkamp, T.M.; Rubbenstroth, D. Monitoring of Free-Ranging and Captive Psittacula Populations in Western Europe for Avian Bornaviruses, Circoviruses and Polyomaviruses. Avian Pathol. 2020, 49, 119–130. [Google Scholar] [CrossRef]
- Morinha, F.; Carrete, M.; Tella, J.L.; Blanco, G. High Prevalence of Novel Beak and Feather Disease Virus in Sympatric Invasive Parakeets Introduced to Spain from Asia and South America. Diversity 2020, 12, 192. [Google Scholar] [CrossRef]
- Kapgate, S.S.; Kumanan, K.; Vijayarani, K.; Barbuddhe, S.B. Avian Parvovirus: Classification, Phylogeny, Pathogenesis and Diagnosis. Avian. Pathol. 2018, 47, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, J.J.; Söderlund-Venermo, M.; Canuti, M.; Eis-Hübinger, A.M.; Hughes, J.; Cotmore, S.F.; Harrach, B. Reorganizing the Family Parvoviridae: A Revised Taxonomy Independent of the Canonical Approach Based on Host Association. Arch. Virol. 2020, 165, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Zádori, Z.; Stefancsik, R.; Rauch, T.; Kisary, J. Analysis of the Complete Nucleotide Sequences of Goose and Muscovy Duck Pervoviruses Indicates Common Ancestral Origin with Adeno-Associated Virus 2. Virology 1995, 212, 562–573. [Google Scholar] [CrossRef]
- Canuti, M.; Verhoeven, J.T.P.; Munro, H.J.; Roul, S.; Ojkic, D.; Robertson, G.J.; Whitney, H.G.; Dufour, S.C.; Lang, A.S. Investigating the Diversity and Host Range of Novel Parvoviruses from North American Ducks Using Epidemiology, Phylogenetics, Genome Structure, and Codon Usage Analysis. Viruses 2021, 13, 193. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.C.; Tomlinson, J.E.; Lopez-Astacio, R.A.; Parrish, C.R.; Van de Walle, G.R. Small but Mighty: Old and New Parvoviruses of Veterinary Significance. Virol. J. 2021, 18, 210. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Zak, R.; Zhang, Y.; Engelhardt, J.F. Inverted Terminal Repeat Sequences Are Important for Intermolecular Recombination and Circularization of Adeno-Associated Virus Genomes. J. Virol. 2005, 79, 364–379. [Google Scholar] [CrossRef]
- Andrews, S. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 7 February 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Tamames, J.; Puente-Sánchez, F. SqueezeMeta, A Highly Portable, Fully Automatic Metagenomic Analysis Pipeline. Front. Microbiol. 2019, 9, 3349. [Google Scholar] [CrossRef]
- Rombel, I.T.; Sykes, K.F.; Rayner, S.; Johnston, S.A. ORF-FINDER: A Vector for High-Throughput Gene Identification. Gene 2002, 282, 33–41. [Google Scholar] [CrossRef]
- Zuker, M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Reese, M.G. Application of a Time-Delay Neural Network to Promoter Annotation in the Drosophila Melanogaster Genome. Comput. Chem. 2001, 26, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Hebsgaard, S.M.; Korning, P.G.; Tolstrup, N.; Engelbrecht, J.; Rouzé, P.; Brunak, S. Splice Site Prediction in Arabidopsis Thaliana Pre-MRNA by Combining Local and Global Sequence Information. Nucleic Acids Res. 1996, 24, 3439–3452. [Google Scholar] [CrossRef]
- Ahmed, F.; Kumar, M.; Raghava, G.P.S. Prediction of Polyadenylation Signals in Human DNA Sequences Using Nucleotide Frequencies. Silico Biol. 2009, 9, 135–148. [Google Scholar] [CrossRef]
- Scott, M.S.; Boisvert, F.-M.; McDowall, M.D.; Lamond, A.I.; Barton, G.J. Characterization and Prediction of Protein Nucleolar Localization Sequences. Nucleic Acids Res. 2010, 38, 7388–7399. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A Computer Program for Analyzing Recombination in, and Removing Signals of Recombination from, Nucleotide Sequence Datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Im, D.S.; Muzyczka, N. The AAV Origin Binding Protein Rep68 Is an ATP-Dependent Site-Specific Endonuclease with DNA Helicase Activity. Cell 1990, 61, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Wang, H.; Wu, H.; Zhang, Q.; Ji, L.; Wang, X.; Shen, Q.; Yang, S.; Ma, X.; Shan, T.; et al. Parvovirus Dark Matter in the Cloaca of Wild Birds. GigaScience 2023, 12, giad001. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.L.; Wonderling, R.S.; Owens, R.A. Mutational Analysis of the Adeno-Associated Virus Type 2 Rep68 Protein Helicase Motifs. J. Virol. 1997, 71, 6996–7004. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, Á.; Mátics, R.; Altan, E.; Delwart, E.; Pankovics, P. A Novel Parvovirus (Family Parvoviridae) in a Freshwater Fish, Zander (Sander lucioperca). Arch. Virol. 2022, 167, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Zádori, Z.; Szelei, J.; Lacoste, M.-C.; Li, Y.; Gariépy, S.; Raymond, P.; Allaire, M.; Nabi, I.R.; Tijssen, P. A Viral Phospholipase A2 Is Required for Parvovirus Infectivity. Dev. Cell 2001, 1, 291–302. [Google Scholar] [CrossRef]
- Carninci, P.; Sandelin, A.; Lenhard, B.; Katayama, S.; Shimokawa, K.; Ponjavic, J.; Semple, C.A.M.; Taylor, M.S.; Engström, P.G.; Frith, M.C.; et al. Genome-Wide Analysis of Mammalian Promoter Architecture and Evolution. Nat. Genet. 2006, 38, 626–635. [Google Scholar] [CrossRef]
- Hartl, D.; Krebs, A.R.; Grand, R.S.; Baubec, T.; Isbel, L.; Wirbelauer, C.; Burger, L.; Schübeler, D. CG Dinucleotides Enhance Promoter Activity Independent of DNA Methylation. Genome Res. 2019, 29, 554–563. [Google Scholar] [CrossRef]
- Shan, T.; Yang, S.; Wang, H.; Wang, H.; Zhang, J.; Gong, G.; Xiao, Y.; Yang, J.; Wang, X.; Lu, J.; et al. Virome in the Cloaca of Wild and Breeding Birds Revealed a Diversity of Significant Viruses. Microbiome 2022, 10, 60. [Google Scholar] [CrossRef]
- de Souza, W.; Romeiro, M.F.; Fumagalli, M.J.; Modha, S.; de Araujo, J.; Queiroz, L.H.; Durigon, E.L.; Figueiredo, L.T.M.; Murcia, P.R.; Gifford, R.J. Chapparvoviruses Occur in at Least Three Vertebrate Classes and Have a Broad Biogeographic Distribution. J. Gen. Virol. 2017, 98, 225–229. [Google Scholar] [CrossRef]
- Vibin, J.; Chamings, A.; Klaassen, M.; Bhatta, T.R.; Alexandersen, S. Metagenomic Characterisation of Avian Parvoviruses and Picornaviruses from Australian Wild Ducks. Sci. Rep. 2020, 10, 12800. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.F.; Fraefel, C.; Seyffert, M. The Interplay between Adeno-Associated Virus and Its Helper Viruses. Viruses 2020, 12, 662. [Google Scholar] [CrossRef] [PubMed]
- Lima, D.A.; Cibulski, S.P.; Finkler, F.; Teixeira, T.F.; Varela, A.P.M.; Cerva, C.; Loiko, M.R.; Scheffer, C.M.; dos Santos, H.F.; Mayer, F.Q.; et al. Faecal Virome of Healthy Chickens Reveals a Large Diversity of the Eukaryote Viral Community, Including Novel Circular SsDNA Viruses. J. Gen. Virol. 2017, 98, 690–703. [Google Scholar] [CrossRef]
- Duarte, M.A.; Silva, J.M.F.; Brito, C.R.; Teixeira, D.S.; Melo, F.L.; Ribeiro, B.M.; Nagata, T.; Campos, F.S. Faecal Virome Analysis of Wild Animals from Brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef] [PubMed]
- Shackelton, L.A.; Hoelzer, K.; Parrish, C.R.; Holmes, E.C. Comparative Analysis Reveals Frequent Recombination in the Parvoviruses. J. Gen. Virol. 2007, 88, 3294–3301. [Google Scholar] [CrossRef]
- Dong, H.V.; Tran, G.T.H.; Nguyen, H.T.T.; Nguyen, T.M.; Trinh, D.Q.; Le, V.P.; Choowongkomon, K.; Rattanasrisomporn, J. Epidemiological Analysis and Genetic Characterization of Parvovirus in Ducks in Northern Vietnam Reveal Evidence of Recombination. Animals 2022, 12, 2846. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Liu, R.; Chen, C.; Cheng, L.; Shi, S.; Fu, G.; Chen, H.; Fu, Q.; Huang, Y. Novel Goose Parvovirus in Domestic Linwu Sheldrakes with Short Beak and Dwarfism Syndrome, China. Transbound. Emerg. Dis. 2019, 66, 1834–1839. [Google Scholar] [CrossRef]
- Wang, J.; Ling, J.; Wang, Z.; Huang, Y.; Zhu, J.; Zhu, G. Molecular Characterization of a Novel Muscovy Duck Parvovirus Isolate: Evidence of Recombination between Classical MDPV and Goose Parvovirus Strains. BMC Vet. Res. 2017, 13, 327. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, Z.; Huang, Y.; Yu, R.; Dong, S.; Li, Z.; Zhang, Y. Identification of a Recombinant Muscovy Duck Parvovirus (MDPV) in Shanghai, China. Vet. Microbiol. 2014, 174, 560–564. [Google Scholar] [CrossRef]
- McWilliam Leitch, E.C.; Cabrerizo, M.; Cardosa, J.; Harvala, H.; Ivanova, O.E.; Kroes, A.C.M.; Lukashev, A.; Muir, P.; Odoom, J.; Roivainen, M.; et al. Evolutionary Dynamics and Temporal/Geographical Correlates of Recombination in the Human Enterovirus Echovirus Types 9, 11, and 30. J. Virol. 2010, 84, 9292–9300. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez, C.; Doménech, A.; Gomez-Lucia, E.; Méndez, J.L.; Ortiz, J.C.; Benítez, L. A Novel Dependoparvovirus Identified in Cloacal Swabs of Monk Parakeet (Myiopsitta monachus) from Urban Areas of Spain. Viruses 2023, 15, 850. https://doi.org/10.3390/v15040850
Sánchez C, Doménech A, Gomez-Lucia E, Méndez JL, Ortiz JC, Benítez L. A Novel Dependoparvovirus Identified in Cloacal Swabs of Monk Parakeet (Myiopsitta monachus) from Urban Areas of Spain. Viruses. 2023; 15(4):850. https://doi.org/10.3390/v15040850
Chicago/Turabian StyleSánchez, Christian, Ana Doménech, Esperanza Gomez-Lucia, José Luis Méndez, Juan Carlos Ortiz, and Laura Benítez. 2023. "A Novel Dependoparvovirus Identified in Cloacal Swabs of Monk Parakeet (Myiopsitta monachus) from Urban Areas of Spain" Viruses 15, no. 4: 850. https://doi.org/10.3390/v15040850
APA StyleSánchez, C., Doménech, A., Gomez-Lucia, E., Méndez, J. L., Ortiz, J. C., & Benítez, L. (2023). A Novel Dependoparvovirus Identified in Cloacal Swabs of Monk Parakeet (Myiopsitta monachus) from Urban Areas of Spain. Viruses, 15(4), 850. https://doi.org/10.3390/v15040850