
Packaging and Uncoating of CRISPR/Cas Ribonucleoproteins for Efficient Gene Editing with Viral and Non-Viral Extracellular Nanoparticles


Abstract
1. Introduction
2. Retroviral Assembly with a Cargo Protein: What Was Known Prior to the CRISPR/Cas Era
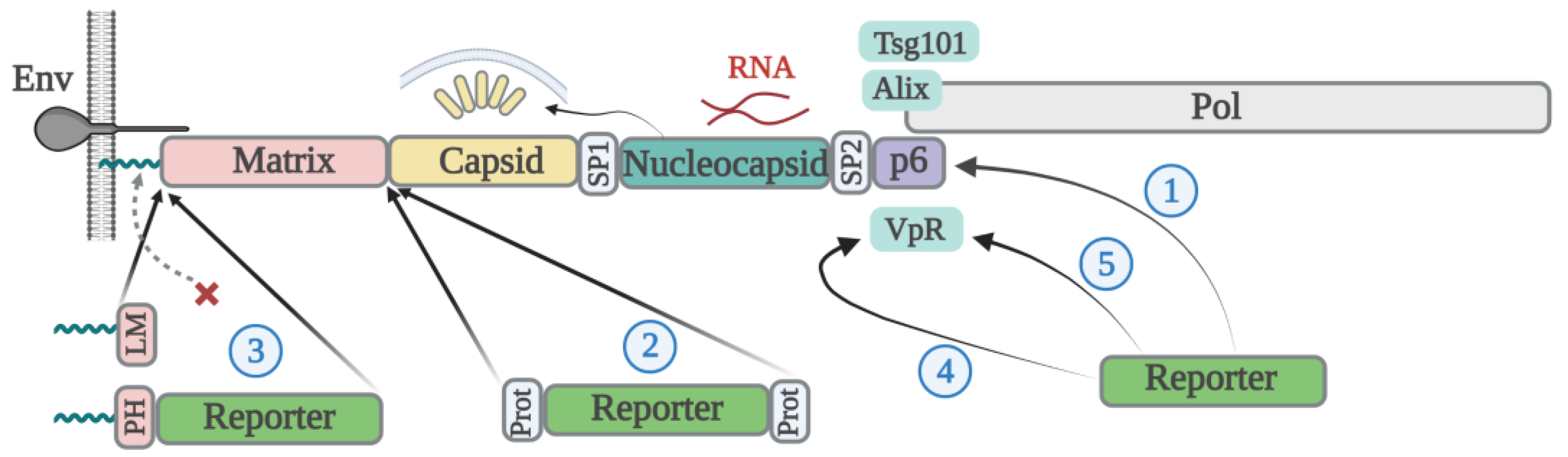
2.1. Lentiviral Particle Assembly
2.2. Packaging Reporter Proteins with Gag
2.3. Vpr-Based Packaging of Reporter Proteins
3. Retroviral Disassembly and Cell Transduction Efficiency with Cargo Proteins
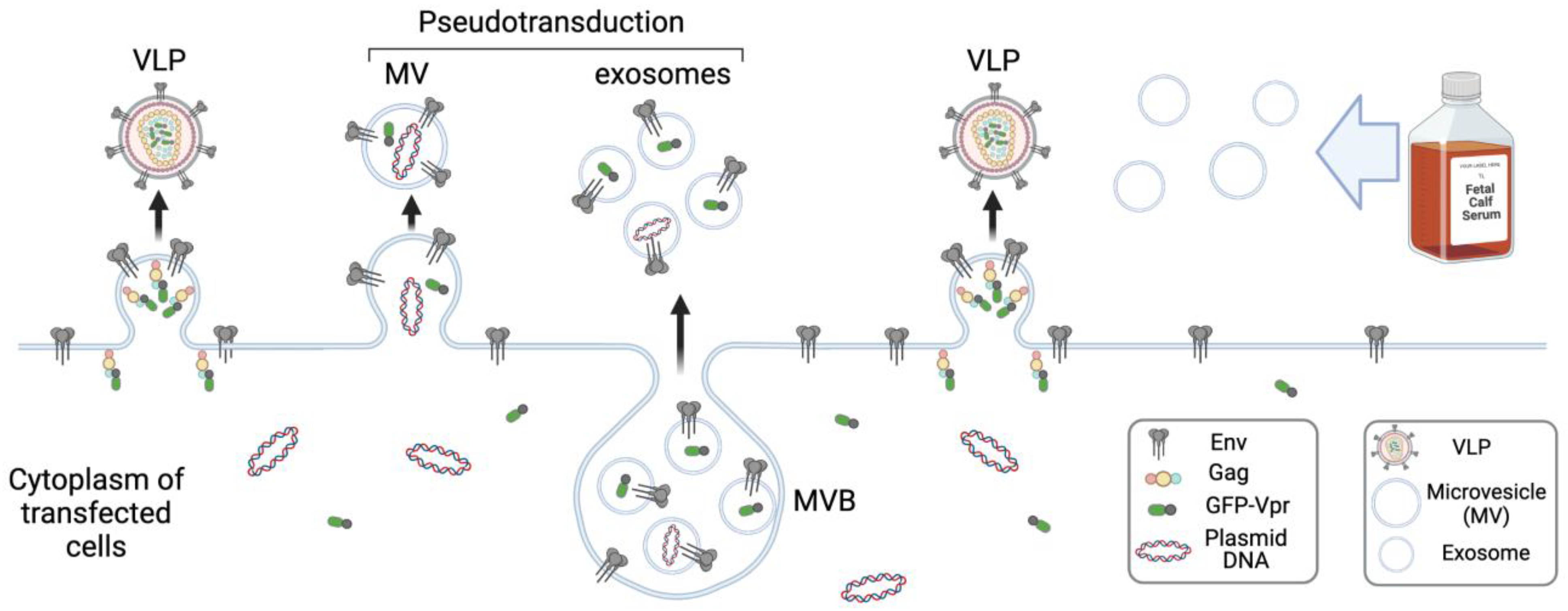
4. Biogenesis and Function of Exosomes
5. Lessons from the Spontaneous and Specific Incorporation of Cas9 RNP into Exosomes
5.1. Nonspecific Loading of EVs with Cas9 RNP
5.2. Specific Mechanisms of Cas9 RNP Recruitment into EVs
5.3. Reversible EV Systems for Packaging and Releasing Cas9 RNP
5.4. Summary
6. Retroviruses for the Delivery of Cas9 RNP: Restrictions and Evolution of Design
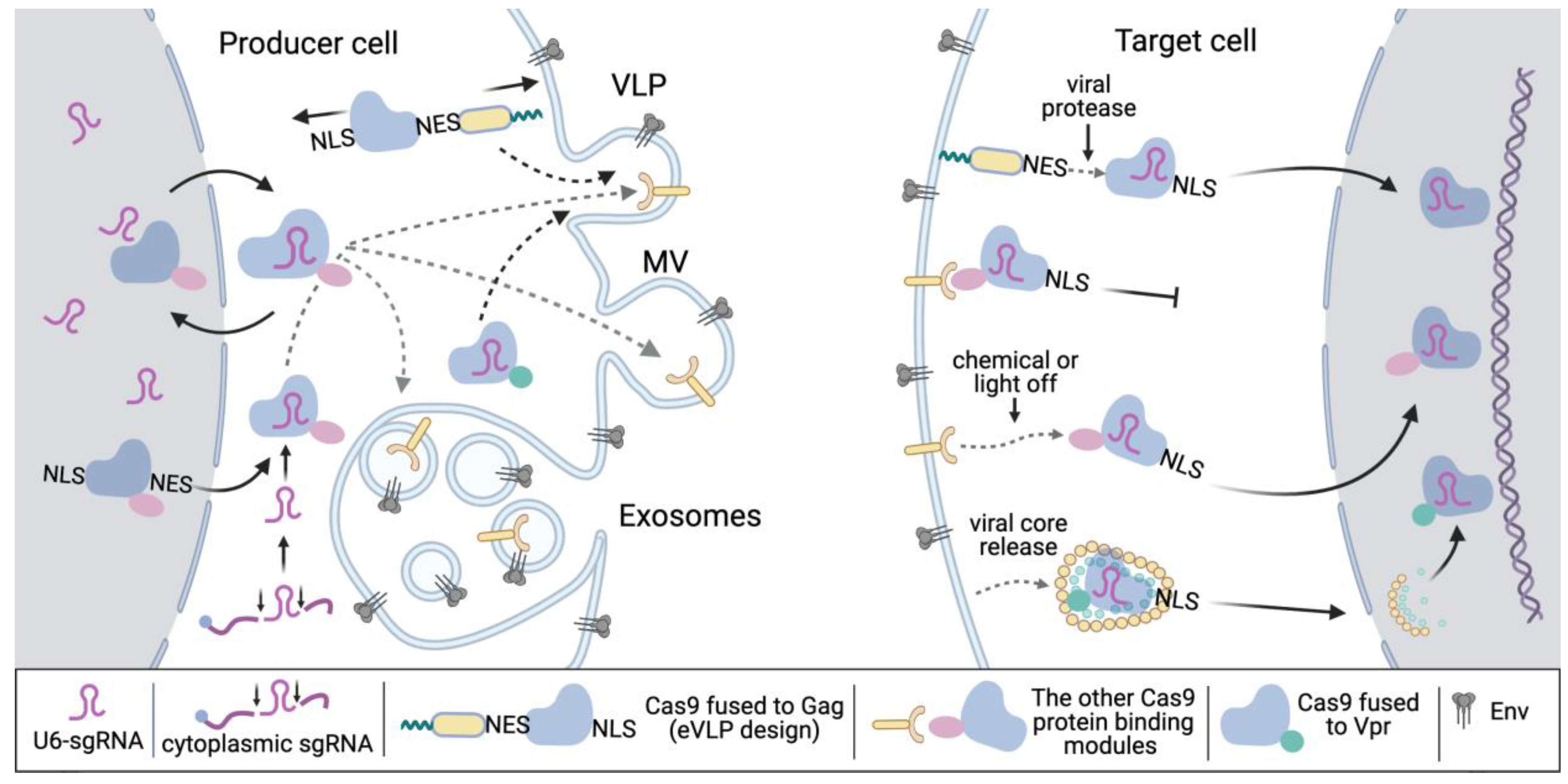
6.1. Fusion of Cas9 with Gag for Delivery with VLPs
6.2. Advanced and Combined Packaging Systems: Dimerization Domains, Vpr, RNA Transduction
6.3. Packaging of Cas9 RNP via sgRNA
6.4. Summary
7. From Particle Assembly to Gene Editing: Important Considerations for Making the System Work Perfectly
7.1. Non-Specific Incorporation
7.2. Stoichiometry
7.3. Temporal Characteristics
7.4. Spatial Issue (sgRNA)
7.5. Spatial Problems (Cas9)
7.6. Size of the Cas Proteins
7.7. Recruiting Mechanism
7.8. Entry
7.9. Disassembly
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Shariat, N.; Dudley, E.G. CRISPRs: Molecular signatures used for pathogen subtyping. Appl. Environ. Microbiol. 2014, 80, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Horodecka, K.; Düchler, M. CRISPR/Cas9: Principle, Applications, and Delivery through Extracellular Vesicles. Int. J. Mol. Sci. 2021, 22, 6072. [Google Scholar] [CrossRef]
- Pausch, P.; Al-Shayeb, B.; Bisom-Rapp, E.; Tsuchida, C.A.; Li, Z.; Cress, B.F.; Knott, G.J.; Jacobsen, S.E.; Banfield, J.F.; Doudna, J.A. CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science 2020, 369, 333–337. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Genovese, P.; Schiroli, G.; Escobar, G.; Tomaso, T.D.; Firrito, C.; Calabria, A.; Moi, D.; Mazzieri, R.; Bonini, C.; Holmes, M.C.; et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 2014, 510, 235–240. [Google Scholar] [CrossRef]
- Azam, B.; Sima, R. Non-Viral Delivery Systems in Gene Therapy and Vaccine Development. In Non-Viral Gene Therapy; Xu-bo, Y., Ed.; IntechOpen: Rijeka, Croatia, 2011. [Google Scholar]
- Mohammadinejad, R.; Dehshahri, A.; Sagar Madamsetty, V.; Zahmatkeshan, M.; Tavakol, S.; Makvandi, P.; Khorsandi, D.; Pardakhty, A.; Ashrafizadeh, M.; Ghasemipour Afshar, E.; et al. In vivo gene delivery mediated by non-viral vectors for cancer therapy. J. Control Release 2020, 325, 249–275. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Piras, F.; Kajaste-Rudnitski, A. Antiviral immunity and nucleic acid sensing in haematopoietic stem cell gene engineering. Gene Ther. 2021, 28, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Yip, B.H. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Cronin, J.; Zhang, X.Y.; Reiser, J. Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther. 2005, 5, 387–398. [Google Scholar] [CrossRef]
- Campbell, S.; Fisher, R.J.; Towler, E.M.; Fox, S.; Issaq, H.J.; Wolfe, T.; Phillips, L.R.; Rein, A. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc. Natl. Acad. Sci. USA 2001, 98, 10875–10879. [Google Scholar] [CrossRef]
- Muriaux, D.; Mirro, J.; Harvin, D.; Rein, A. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. USA 2001, 98, 5246–5251. [Google Scholar] [CrossRef] [PubMed]
- Rulli, S.J., Jr.; Hibbert, C.S.; Mirro, J.; Pederson, T.; Biswal, S.; Rein, A. Selective and nonselective packaging of cellular RNAs in retrovirus particles. J. Virol. 2007, 81, 6623–6631. [Google Scholar] [CrossRef]
- Bryant, M.; Ratner, L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. USA 1990, 87, 523–527. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Luukkonen, B.G.; Fenyö, E.M.; Schwartz, S. Overexpression of human immunodeficiency virus type 1 protease increases intracellular cleavage of Gag and reduces virus infectivity. Virology 1995, 206, 854–865. [Google Scholar] [CrossRef]
- Shehu-Xhilaga, M.; Crowe, S.M.; Mak, J. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 2001, 75, 1834–1841. [Google Scholar] [CrossRef]
- Westerman, K.A.; Ao, Z.; Cohen, E.A.; Leboulch, P. Design of a trans protease lentiviral packaging system that produces high titer virus. Retrovirology 2007, 4, 96. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ablan, S.; Freed, E.O.; Tanaka, Y. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J. Virol. 2004, 78, 1026–1031. [Google Scholar] [CrossRef]
- Wyma, D.J.; Jiang, J.; Shi, J.; Zhou, J.; Lineberger, J.E.; Miller, M.D.; Aiken, C. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: A novel role of the gp41 cytoplasmic tail. J. Virol. 2004, 78, 3429–3435. [Google Scholar] [CrossRef] [PubMed]
- Hermida-Matsumoto, L.; Resh, M.D. Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imaging. J. Virol. 2000, 74, 8670–8679. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Tricaud, C.; Davoust, J.; Jones, I.M. Tagging the human immunodeficiency virus gag protein with green fluorescent protein. Minimal evidence for colocalisation with actin. Virology 1999, 255, 20–25. [Google Scholar] [CrossRef]
- Pornillos, O.; Higginson, D.S.; Stray, K.M.; Fisher, R.D.; Garrus, J.E.; Payne, M.; He, G.P.; Wang, H.E.; Morham, S.G.; Sundquist, W.I. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J. Cell Biol. 2003, 162, 425–434. [Google Scholar] [CrossRef]
- Müller, B.; Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Zentgraf, H.; Kräusslich, H.G. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J. Virol. 2004, 78, 10803–10813. [Google Scholar] [CrossRef]
- Hübner, W.; Chen, P.; Del Portillo, A.; Liu, Y.; Gordon, R.E.; Chen, B.K. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J. Virol. 2007, 81, 12596–12607. [Google Scholar] [CrossRef]
- Hübner, W.; McNerney, G.P.; Chen, P.; Dale, B.M.; Gordon, R.E.; Chuang, F.Y.; Li, X.D.; Asmuth, D.M.; Huser, T.; Chen, B.K. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 2009, 323, 1743–1747. [Google Scholar] [CrossRef]
- Real, F.; Sennepin, A.; Ganor, Y.; Schmitt, A.; Bomsel, M. Live Imaging of HIV-1 Transfer across T Cell Virological Synapse to Epithelial Cells that Promotes Stromal Macrophage Infection. Cell Rep. 2018, 23, 1794–1805. [Google Scholar] [CrossRef]
- Voelkel, C.; Galla, M.; Maetzig, T.; Warlich, E.; Kuehle, J.; Zychlinski, D.; Bode, J.; Cantz, T.; Schambach, A.; Baum, C. Protein transduction from retroviral Gag precursors. Proc. Natl. Acad. Sci. USA 2010, 107, 7805–7810. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, B.; Musier-Forsyth, K.; Mansky, L.M.; Mueller, J.D. Fluorescence fluctuation spectroscopy on viral-like particles reveals variable gag stoichiometry. Biophys J. 2009, 96, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Urano, E.; Aoki, T.; Futahashi, Y.; Murakami, T.; Morikawa, Y.; Yamamoto, N.; Komano, J. Substitution of the myristoylation signal of human immunodeficiency virus type 1 Pr55Gag with the phospholipase C-delta1 pleckstrin homology domain results in infectious pseudovirion production. J. Gen. Virol. 2008, 89 Pt 12, 3144–3149. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Shimizu, S.; Urano, E.; Futahashi, Y.; Hamatake, M.; Tamamura, H.; Terashima, K.; Murakami, T.; Yamamoto, N.; Komano, J. Improvement of lentiviral vector-mediated gene transduction by genetic engineering of the structural protein Pr55 Gag. Gene Ther. 2010, 17, 1124–1133. [Google Scholar] [CrossRef]
- Aoki, T.; Miyauchi, K.; Urano, E.; Ichikawa, R.; Komano, J. Protein transduction by pseudotyped lentivirus-like nanoparticles. Gene Ther. 2011, 18, 936–941. [Google Scholar] [CrossRef]
- Bachand, F.; Yao, X.J.; Hrimech, M.; Rougeau, N.; Cohen, E.A. Incorporation of Vpr into human immunodeficiency virus type 1 requires a direct interaction with the p6 domain of the p55 gag precursor. J. Biol. Chem. 1999, 274, 9083–9091. [Google Scholar] [CrossRef]
- Wanaguru, M.; Bishop, K.N. HIV-1 Gag Recruits Oligomeric Vpr via Two Binding Sites in p6, but Both Mature p6 and Vpr Are Rapidly Lost upon Target Cell Entry. J. Virol. 2021, 95, e0055421. [Google Scholar] [CrossRef]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef]
- Muthumani, K.; Montaner, L.J.; Ayyavoo, V.; Weiner, D.B. Vpr-GFP virion particle identifies HIV-infected targets and preserves HIV-1Vpr function in macrophages and T-cells. DNA Cell Biol. 2000, 19, 179–188. [Google Scholar] [CrossRef]
- Jenkins, Y.; Pornillos, O.; Rich, R.L.; Myszka, D.G.; Sundquist, W.I.; Malim, M.H. Biochemical analyses of the interactions between human immunodeficiency virus type 1 Vpr and p6(Gag). J. Virol. 2001, 75, 10537–10542. [Google Scholar] [CrossRef]
- Cavrois, M.; De Noronha, C.; Greene, W.C. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 2002, 20, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Accola, M.A.; Bukovsky, A.A.; Jones, M.S.; Göttlinger, H.G. A conserved dileucine-containing motif in p6(gag) governs the particle association of Vpx and Vpr of simian immunodeficiency viruses SIV(mac) and SIV(agm). J. Virol. 1999, 73, 9992–9999. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Tessmer, U.; Schubert, U.; Kräusslich, H.G. Human immunodeficiency virus type 1 Vpr protein is incorporated into the virion in significantly smaller amounts than gag and is phosphorylated in infected cells. J. Virol. 2000, 74, 9727–9731. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.G.; Zila, V.; Peters, K.; Schifferdecker, S.; Stanic, M.; Lucic, B.; Laketa, V.; Lusic, M.; Müller, B.; Kräusslich, H.G. HIV-1 uncoating by release of viral cDNA from capsid-like structures in the nucleus of infected cells. Elife 2021, 10, e64776. [Google Scholar] [CrossRef]
- Li, C.; Burdick, R.C.; Nagashima, K.; Hu, W.S.; Pathak, V.K. HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc. Natl. Acad. Sci. USA 2021, 118, e2019467118. [Google Scholar] [CrossRef]
- Murriel, C.L.; Dowdy, S.F. Influence of protein transduction domains on intracellular delivery of macromolecules. Expert Opin. Drug Deliv. 2006, 3, 739–746. [Google Scholar] [CrossRef]
- Kaczmarczyk, S.J.; Sitaraman, K.; Young, H.A.; Hughes, S.H.; Chatterjee, D.K. Protein delivery using engineered virus-like particles. Proc. Natl. Acad. Sci. USA 2011, 108, 16998–17003. [Google Scholar] [CrossRef]
- Desai, T.M.; Marin, M.; Sood, C.; Shi, J.; Nawaz, F.; Aiken, C.; Melikyan, G.B. Fluorescent protein-tagged Vpr dissociates from HIV-1 core after viral fusion and rapidly enters the cell nucleus. Retrovirology 2015, 12, 88. [Google Scholar] [CrossRef]
- Jenkins, Y.; McEntee, M.; Weis, K.; Greene, W.C. Characterization of HIV-1 vpr nuclear import: Analysis of signals and pathways. J. Cell Biol. 1998, 143, 875–885. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, X.; Barnes, C.O.; DeLucia, M.; Cohen, A.E.; Gronenborn, A.M.; Ahn, J.; Calero, G. The DDB1-DCAF1-Vpr-UNG2 crystal structure reveals how HIV-1 Vpr steers human UNG2 toward destruction. Nat. Struct Mol. Biol. 2016, 23, 933–940. [Google Scholar] [CrossRef]
- Wen, X.; Duus, K.M.; Friedrich, T.D.; de Noronha, C.M.C. The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VprBP/DCAF1 as an adaptor. J. Biol. Chem. 2007, 282, 27046–27057. [Google Scholar] [CrossRef] [PubMed]
- Reuschl, A.K.; Mesner, D.; Shivkumar, M.; Whelan, M.V.X.; Pallett, L.J.; Guerra-Assunção, J.A.; Madansein, R.; Dullabh, K.J.; Sigal, A.; Thornhill, J.P.; et al. HIV-1 Vpr drives a tissue residency-like phenotype during selective infection of resting memory T cells. Cell Rep. 2022, 39, 110650. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Parra, S.; Marin, M.; Gahlaut, N.; Suter, R.; Kondo, N.; Melikyan, G.B. Fusion of mature HIV-1 particles leads to complete release of a gag-GFP-based content marker and raises the intraviral pH. PLoS ONE 2013, 8, e71002. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Teis, D. The ESCRT machinery. Curr. Biol. 2012, 22, R116–R120. [Google Scholar] [CrossRef] [PubMed]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Göttlinger, H.G. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- Rezaie, J.; Aslan, C.; Ahmadi, M.; Zolbanin, N.M.; Kashanchi, F.; Jafari, R. The versatile role of exosomes in human retroviral infections: From immunopathogenesis to clinical application. Cell Biosci. 2021, 11, 19. [Google Scholar] [CrossRef]
- Sung, B.H.; von Lersner, A.; Guerrero, J.; Krystofiak, E.S.; Inman, D.; Pelletier, R.; Zijlstra, A.; Ponik, S.M.; Weaver, A.M. A live cell reporter of exosome secretion and uptake reveals pathfinding behavior of migrating cells. Nat. Commun. 2020, 11, 2092. [Google Scholar] [CrossRef]
- Reiter, K.; Aguilar, P.P.; Wetter, V.; Steppert, P.; Tover, A.; Jungbauer, A. Separation of virus-like particles and extracellular vesicles by flow-through and heparin affinity chromatography. J. Chromatogr. A 2019, 1588, 77–84. [Google Scholar] [CrossRef]
- Van Engelenburg, S.B.; Shtengel, G.; Sengupta, P.; Waki, K.; Jarnik, M.; Ablan, S.D.; Freed, E.O.; Hess, H.F.; Lippincott-Schwartz, J. Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science 2014, 343, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Pelchen-Matthews, A.; Kramer, B.; Marsh, M. Infectious HIV-1 assembles in late endosomes in primary macrophages. J. Cell Biol. 2003, 162, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Lavado-García, J.; González-Domínguez, I.; Cervera, L.; Jorge, I.; Vázquez, J.; Gòdia, F. Molecular Characterization of the Coproduced Extracellular Vesicles in HEK293 during Virus-Like Particle Production. J. Proteome Res. 2020, 19, 4516–4532. [Google Scholar] [CrossRef] [PubMed]
- Mangeot, P.E.; Dollet, S.; Girard, M.; Ciancia, C.; Joly, S.; Peschanski, M.; Lotteau, V. Protein transfer into human cells by VSV-G-induced nanovesicles. Mol. Ther. 2011, 19, 1656–1666. [Google Scholar] [CrossRef]
- Aronheim, A.; Engelberg, D.; Li, N.; al-Alawi, N.; Schlessinger, J.; Karin, M. Membrane targeting of the nucleotide exchange factor Sos is sufficient for activating the Ras signaling pathway. Cell 1994, 78, 949–961. [Google Scholar] [CrossRef]
- Liu, M.L.; Winther, B.L.; Kay, M.A. Pseudotransduction of hepatocytes by using concentrated pseudotyped vesicular stomatitis virus G glycoprotein (VSV-G)-Moloney murine leukemia virus-derived retrovirus vectors: Comparison of VSV-G and amphotropic vectors for hepatic gene transfer. J. Virol. 1996, 70, 2497–2502. [Google Scholar] [CrossRef]
- Gallardo, H.F.; Tan, C.; Ory, D.; Sadelain, M. Recombinant retroviruses pseudotyped with the vesicular stomatitis virus G glycoprotein mediate both stable gene transfer and pseudotransduction in human peripheral blood lymphocytes. Blood 1997, 90, 952–957. [Google Scholar] [CrossRef]
- Montagna, C.; Petris, G.; Casini, A.; Maule, G.; Franceschini, G.M.; Zanella, I.; Conti, L.; Arnoldi, F.; Burrone, O.R.; Zentilin, L.; et al. VSV-G-Enveloped Vesicles for Traceless Delivery of CRISPR-Cas9. Mol. Ther. Nucleic. Acids 2018, 12, 453–462. [Google Scholar] [CrossRef]
- Chen, R.; Huang, H.; Liu, H.; Xi, J.; Ning, J.; Zeng, W.; Shen, C.; Zhang, T.; Yu, G.; Xu, Q.; et al. Friend or Foe? Evidence Indicates Endogenous Exosomes Can Deliver Functional gRNA and Cas9 Protein. Small 2019, 15, e1902686. [Google Scholar] [CrossRef]
- Mazurov, D.; Ilinskaya, A.; Heidecker, G.; Lloyd, P.; Derse, D. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. 2010, 6, e1000788. [Google Scholar] [CrossRef]
- Shunaeva, A.; Potashnikova, D.; Pichugin, A.; Mishina, A.; Filatov, A.; Nikolaitchik, O.; Hu, W.S.; Mazurov, D. Improvement of HIV-1 and Human T Cell Lymphotropic Virus Type 1 Replication-Dependent Vectors via Optimization of Reporter Gene Reconstitution and Modification with Intronic Short Hairpin RNA. J. Virol. 2015, 89, 10591–10601. [Google Scholar] [CrossRef] [PubMed]
- Lainšček, D.; Kadunc, L.; Keber, M.M.; Bratkovič, I.H.; Romih, R.; Jerala, R. Delivery of an Artificial Transcription Regulator dCas9-VPR by Extracellular Vesicles for Therapeutic Gene Activation. ACS Synth Biol. 2018, 7, 2715–2725. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, J.; Kadungure, T.; Beyene, J.; Zhang, H.; Lu, Q. ARMMs as a versatile platform for intracellular delivery of macromolecules. Nat. Commun. 2018, 9, 960. [Google Scholar] [CrossRef]
- Kubala, M.H.; Kovtun, O.; Alexandrov, K.; Collins, B.M. Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci. 2010, 19, 2389–2401. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, X.; Xie, F.; Xu, B.; Xie, P.; Yang, T.; Shi, Q.; Zhang, C.Y.; Zhang, Y.; Chen, J.; et al. An engineered exosome for delivering sgRNA:Cas9 ribonucleoprotein complex and genome editing in recipient cells. Biomater Sci. 2020, 8, 2966–2976. [Google Scholar] [CrossRef]
- Ye, Y.; Shi, Q.; Yang, T.; Xie, F.; Zhang, X.; Xu, B.; Fang, J.; Chen, J.; Zhang, Y.; Li, J. In Vivo Visualized Tracking of Tumor-Derived Extracellular Vesicles Using CRISPR-Cas9 System. Technol. Cancer Res. Treat 2022, 21, 15330338221085370. [Google Scholar] [CrossRef]
- Igawa, T.; Ishii, S.; Tachibana, T.; Maeda, A.; Higuchi, Y.; Shimaoka, S.; Moriyama, C.; Watanabe, T.; Takubo, R.; Doi, Y.; et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat. Biotechnol. 2010, 28, 1203–1207. [Google Scholar] [CrossRef]
- Yao, X.; Lyu, P.; Yoo, K.; Yadav, M.K.; Singh, R.; Atala, A.; Lu, B. Engineered extracellular vesicles as versatile ribonucleoprotein delivery vehicles for efficient and safe CRISPR genome editing. J. Extracell Vesicles 2021, 10, e12076. [Google Scholar] [CrossRef]
- Campbell, L.A.; Coke, L.M.; Richie, C.T.; Fortuno, L.V.; Park, A.Y.; Harvey, B.K. Gesicle-Mediated Delivery of CRISPR/Cas9 Ribonucleoprotein Complex for Inactivating the HIV Provirus. Mol. Ther. 2019, 27, 151–163. [Google Scholar] [CrossRef]
- Osteikoetxea, X.; Silva, A.; Lázaro-Ibáñez, E.; Salmond, N.; Shatnyeva, O.; Stein, J.; Schick, J.; Wren, S.; Lindgren, J.; Firth, M.; et al. Engineered Cas9 extracellular vesicles as a novel gene editing tool. J. Extracell Vesicles 2022, 11, e12225. [Google Scholar] [CrossRef] [PubMed]
- Florkiewicz, R.Z.; Rose, J.K. A cell line expressing vesicular stomatitis virus glycoprotein fuses at low pH. Science 1984, 225, 721–723. [Google Scholar] [CrossRef] [PubMed]
- Kanada, M.; Bachmann, M.H.; Hardy, J.W.; Frimannson, D.O.; Bronsart, L.; Wang, A.; Sylvester, M.D.; Schmidt, T.L.; Kaspar, R.L.; Butte, M.J.; et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. USA 2015, 112, E1433–E1442. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.E.; Leonard, J.N. A platform for actively loading cargo RNA to elucidate limiting steps in EV-mediated delivery. J. Extracell Vesicles 2016, 5, 31027. [Google Scholar] [CrossRef]
- Kojima, R.; Bojar, D.; Rizzi, G.; Hamri, G.C.; El-Baba, M.D.; Saxena, P.; Ausländer, S.; Tan, K.R.; Fussenegger, M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 2018, 9, 1305. [Google Scholar] [CrossRef]
- Belmont, B.J.; Niles, J.C. Engineering a direct and inducible protein-RNA interaction to regulate RNA biology. ACS Chem. Biol. 2010, 5, 851–861. [Google Scholar] [CrossRef]
- Choi, J.G.; Dang, Y.; Abraham, S.; Ma, H.; Zhang, J.; Guo, H.; Cai, Y.; Mikkelsen, J.G.; Wu, H.; Shankar, P.; et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther. 2016, 23, 627–633. [Google Scholar] [CrossRef]
- Accola, M.A.; Strack, B.; Göttlinger, H.G. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J. Virol. 2000, 74, 5395–5402. [Google Scholar] [CrossRef]
- Mangeot, P.E.; Risson, V.; Fusil, F.; Marnef, A.; Laurent, E.; Blin, J.; Mournetas, V.; Massouridès, E.; Sohier, T.J.M.; Corbin, A.; et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019, 10, 45. [Google Scholar] [CrossRef]
- Amirache, F.; Lévy, C.; Costa, C.; Mangeot, P.E.; Torbett, B.E.; Wang, C.X.; Nègre, D.; Cosset, F.L.; Verhoeyen, E. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood 2014, 123, 1422–1424. [Google Scholar] [CrossRef]
- Girard-Gagnepain, A.; Amirache, F.; Costa, C.; Lévy, C.; Frecha, C.; Fusil, F.; Nègre, D.; Lavillette, D.; Cosset, F.L.; Verhoeyen, E. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood 2014, 124, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Gee, P.; Lung, M.S.Y.; Okuzaki, Y.; Sasakawa, N.; Iguchi, T.; Makita, Y.; Hozumi, H.; Miura, Y.; Yang, L.F.; Iwasaki, M.; et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat. Commun. 2020, 11, 1334. [Google Scholar] [CrossRef] [PubMed]
- Chowrira, B.M.; Pavco, P.A.; McSwiggen, J.A. In vitro and in vivo comparison of hammerhead, hairpin, and hepatitis delta virus self-processing ribozyme cassettes. J. Biol. Chem. 1994, 269, 25856–25864. [Google Scholar] [CrossRef]
- Ma, H.; Tu, L.C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. CRISPR-Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol. 2016, 214, 529–537. [Google Scholar] [CrossRef]
- Hamilton, J.R.; Tsuchida, C.A.; Nguyen, D.N.; Shy, B.R.; McGarrigle, E.R.; Sandoval Espinoza, C.R.; Carr, D.; Blaeschke, F.; Marson, A.; Doudna, J.A. Targeted delivery of CRISPR-Cas9 and transgenes enables complex immune cell engineering. Cell Rep. 2021, 35, 109207. [Google Scholar] [CrossRef] [PubMed]
- Indikova, I.; Indik, S. Highly efficient ’hit-and-run’ genome editing with unconcentrated lentivectors carrying Vpr.Prot.Cas9 protein produced from RRE-containing transcripts. Nucleic Acids Res. 2020, 48, 8178–8187. [Google Scholar] [CrossRef]
- Pocock, G.M.; Becker, J.T.; Swanson, C.M.; Ahlquist, P.; Sherer, N.M. HIV-1 and M-PMV RNA Nuclear Export Elements Program Viral Genomes for Distinct Cytoplasmic Trafficking Behaviors. PLoS Pathog. 2016, 12, e1005565. [Google Scholar] [CrossRef]
- Selig, L.; Pages, J.C.; Tanchou, V.; Prévéral, S.; Berlioz-Torrent, C.; Liu, L.X.; Erdtmann, L.; Darlix, J.; Benarous, R.; Benichou, S. Interaction with the p6 domain of the gag precursor mediates incorporation into virions of Vpr and Vpx proteins from primate lentiviruses. J. Virol. 1999, 73, 592–600. [Google Scholar] [CrossRef]
- Lu, B.; Javidi-Parsijani, P.; Makani, V.; Mehraein-Ghomi, F.; Sarhan, W.M.; Sun, D.; Yoo, K.W.; Atala, Z.P.; Lyu, P.; Atala, A. Delivering SaCas9 mRNA by lentivirus-like bionanoparticles for transient expression and efficient genome editing. Nucleic Acids Res. 2019, 47, e44. [Google Scholar] [CrossRef]
- Lyu, P.; Javidi-Parsijani, P.; Atala, A.; Lu, B. Delivering Cas9/sgRNA ribonucleoprotein (RNP) by lentiviral capsid-based bionanoparticles for efficient ’hit-and-run’ genome editing. Nucleic Acids Res. 2019, 47, e99. [Google Scholar] [CrossRef]
- Banskota, S.; Raguram, A.; Suh, S.; Du, S.W.; Davis, J.R.; Choi, E.H.; Wang, X.; Nielsen, S.C.; Newby, G.A.; Randolph, P.B.; et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell 2022, 185, 250–265.e16. [Google Scholar] [CrossRef] [PubMed]
- Knapp, D.; Michaels, Y.S.; Jamilly, M.; Ferry, Q.R.V.; Barbosa, H.; Milne, T.A.; Fulga, T.A. Decoupling tRNA promoter and processing activities enables specific Pol-II Cas9 guide RNA expression. Nat. Commun. 2019, 10, 1490. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Sun, W.; Yin, W.; Ma, L.; Liu, Y. Rational design of Cas9 ribonucleoprotein with a “gRNA-shRNA” for multidimensional genome manipulation and enhanced homology-directed repair. bioRxiv 2022. [Google Scholar] [CrossRef]
- Campa, C.C.; Weisbach, N.R.; Santinha, A.J.; Incarnato, D.; Platt, R.J. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 2019, 16, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, J.P.; Rios, A.R.; Wu, L.; Qi, L.S. Enhanced Cas12a multi-gene regulation using a CRISPR array separator. Elife 2021, 10, e66406. [Google Scholar] [CrossRef]
- Niopek, D.; Benzinger, D.; Roensch, J.; Draebing, T.; Wehler, P.; Eils, R.; Di Ventura, B. Engineering light-inducible nuclear localization signals for precise spatiotemporal control of protein dynamics in living cells. Nat. Commun. 2014, 5, 4404. [Google Scholar] [CrossRef]
- Niopek, D.; Wehler, P.; Roensch, J.; Eils, R.; Di Ventura, B. Optogenetic control of nuclear protein export. Nat. Commun. 2016, 7, 10624. [Google Scholar] [CrossRef] [PubMed]
- Di Ventura, B.; Kuhlman, B. Go in! Go out! Inducible control of nuclear localization. Curr. Opin. Chem. Biol. 2016, 34, 62–71. [Google Scholar] [CrossRef]
- Liu, K.I.; Ramli, M.N.; Woo, C.W.; Wang, Y.; Zhao, T.; Zhang, X.; Yim, G.R.; Chong, B.Y.; Gowher, A.; Chua, M.Z.; et al. A chemical-inducible CRISPR-Cas9 system for rapid control of genome editing. Nat. Chem. Biol. 2016, 12, 980–987. [Google Scholar] [CrossRef]
- Chen, S.; Liu, Z.; Xie, W.; Yu, H.; Lai, L.; Li, Z. Compact Cje3Cas9 for Efficient In Vivo Genome Editing and Adenine Base Editing. Crispr. J. 2022, 5, 472–486. [Google Scholar] [CrossRef]
- Pausch, P.; Soczek, K.M.; Herbst, D.A.; Tsuchida, C.A.; Al-Shayeb, B.; Banfield, J.F.; Nogales, E.; Doudna, J.A. DNA interference states of the hypercompact CRISPR-CasΦ effector. Nat. Struct Mol. Biol. 2021, 28, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Lee, J.M.; Moon, S.B.; Chin, H.J.; Park, S.; Lim, Y.; Kim, D.; Koo, T.; Ko, J.H.; Kim, Y.S. Efficient CRISPR editing with a hypercompact Cas12f1 and engineered guide RNAs delivered by adeno-associated virus. Nat. Biotechnol. 2022, 40, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chemparathy, A.; Zeng, L.; Kempton, H.R.; Shang, S.; Nakamura, M.; Qi, L.S. Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Mol. Cell 2021, 81, 4333–4345.e4. [Google Scholar] [CrossRef] [PubMed]
- Xin, C.; Yin, J.; Yuan, S.; Ou, L.; Liu, M.; Zhang, W.; Hu, J. Comprehensive assessment of miniature CRISPR-Cas12f nucleases for gene disruption. Nat. Commun. 2022, 13, 5623. [Google Scholar] [CrossRef]
- Segel, M.; Lash, B.; Song, J.; Ladha, A.; Liu, C.C.; Jin, X.; Mekhedov, S.L.; Macrae, R.K.; Koonin, E.V.; Zhang, F. Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science 2021, 373, 882–889. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Mechanism of Assembly | Mechanism of Disassembly | Features | Ref. |
|---|---|---|---|---|
| 1. VEsiCas | Absent | Spontaneous release | No VSVG, sgRNA under T7 promoter | [69] |
| 2. GEDEX | Absent | Spontaneous release | No VSVG, U6-sgRNA | [73] |
| 3. ARMMs | ARRDC1 + WW-Cas9 | Absent | No VSVG, U6-sgRNA | [75] |
| 4. CD63 | CD63-GFP + Cas9-anti-GFP-nanobody | Absent. Acidification in late endosomes? | No VSVG, U6-sgRNA, no CD63+ exosome isolation | [77,78] |
| 5. Aptamer com | Com-CD63-Com + sgRNA-com | Absent | VSVG, U6-sgRNA, Cas9 recruitment via sgRNA | [80] |
| 6. Cherry Picker Red | CherryPicker-FRBP12 + Cas9-FRB, chemical | Removal of chemical dimerizer | VSVG, U6-sgRNA, no CherryPicker+ exosome isolation | [81] |
| 7. CIBN-CRY2 | Palm(Myr)-CIBN (CD9-CIBN) + Cas9-CRY2, light | Turning off the light | No VSVG, U6-sgRNA, comparative study | [82] |
| Name | Mechanism of Assembly | Mechanism of Disassembly | Features | Ref. |
|---|---|---|---|---|
| 1. PH-Cas9-GagPol | Fusion to the N-terminus of HIV GagPol | Cleavage by HIV protease | Lentiviral U6-sgRNA delivery to target cells | [88] |
| 2. Gag-Cas9, MiniGag-Cas9 | Fusion to the C-terminus of HIV Gag | Co-delivery of Cas9 to VLPs and EVs, U6-sgRNA | [89] | |
| 3. Nanoblades | MLV Gag-Cas9 fusion | Cleavage by MLV protease, low level | BaEV Env, U6-sgRNA | [90] |
| 4. NanoMEDIC | LM-FKBP12-HIV-Gag + FRB-Cas9 (chemical) and LTR-Ψ-HH-sgRNA-HDV (RNA) | Removal of chemical dimerizer AP21967 | Comparative study, double recruiting mechanism, not with HIV Env | [93] |
| 5. HIV Gag-Cas9 | Fusion to the C-terminus of HIV Gag | Cleavage by HIV protease | U6-sgRNA, CAR transduction, delivery to CD4 lymphocytes | [96] |
| 6. Vpr-Prot-Cas9 | p6-Vpr interaction and protease cleavage | Cleavage by HIV protease + core release | Parallel lentiviral transduction,no packaging of sgRNA | [97] |
| 7. Aptamer com | NC-Com + sgRNA-com (RNA) | U6-sgRNA, Cas9 recruitment via sgRNA | [101] | |
| 8. eVLP | MLV Gag-Cas9 fusion + NES | Cleavage by MLV protease, optimized | U6-sgRNA, cleavable NES | [102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazurov, D.; Ramadan, L.; Kruglova, N. Packaging and Uncoating of CRISPR/Cas Ribonucleoproteins for Efficient Gene Editing with Viral and Non-Viral Extracellular Nanoparticles. Viruses 2023, 15, 690. https://doi.org/10.3390/v15030690
Mazurov D, Ramadan L, Kruglova N. Packaging and Uncoating of CRISPR/Cas Ribonucleoproteins for Efficient Gene Editing with Viral and Non-Viral Extracellular Nanoparticles. Viruses. 2023; 15(3):690. https://doi.org/10.3390/v15030690
Chicago/Turabian StyleMazurov, Dmitriy, Lama Ramadan, and Natalia Kruglova. 2023. "Packaging and Uncoating of CRISPR/Cas Ribonucleoproteins for Efficient Gene Editing with Viral and Non-Viral Extracellular Nanoparticles" Viruses 15, no. 3: 690. https://doi.org/10.3390/v15030690
APA StyleMazurov, D., Ramadan, L., & Kruglova, N. (2023). Packaging and Uncoating of CRISPR/Cas Ribonucleoproteins for Efficient Gene Editing with Viral and Non-Viral Extracellular Nanoparticles. Viruses, 15(3), 690. https://doi.org/10.3390/v15030690