

Genetic Diversity of Norovirus in Children with Acute Gastroenteritis in Southwest Nigeria, 2015–2017

 , , and
, , and 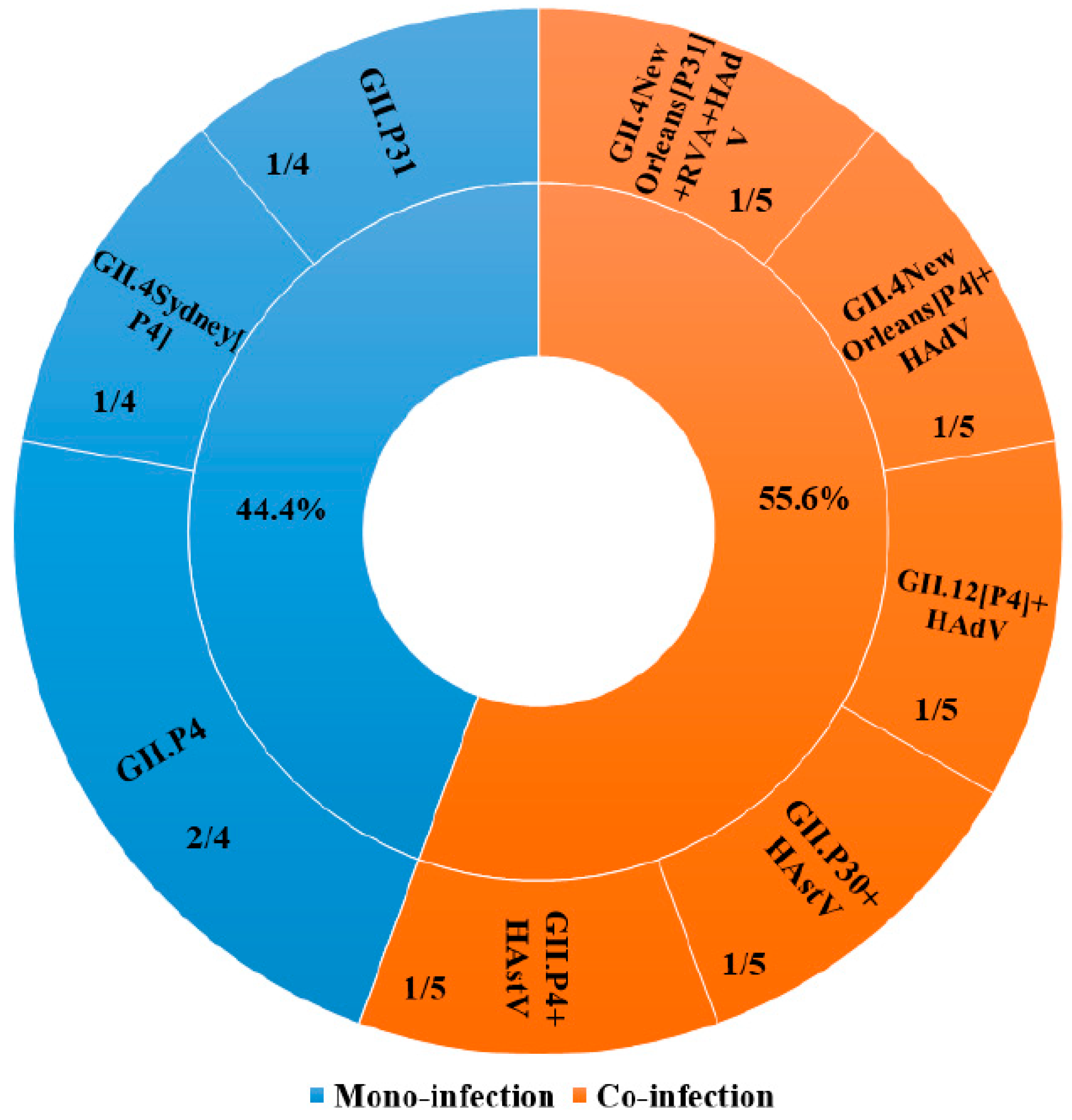{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Sample Collection
2.3. Detection of NoV
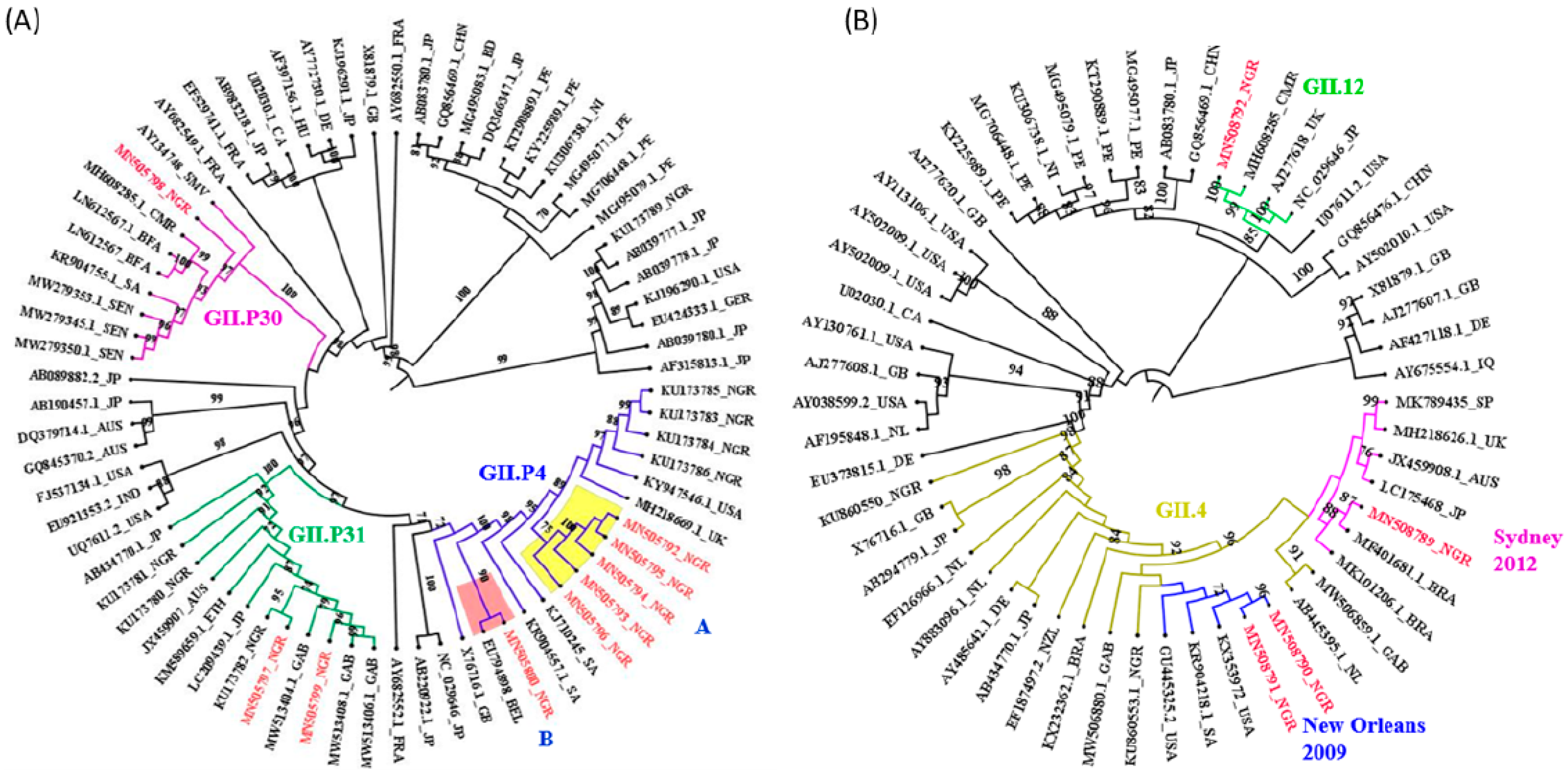
2.4. Nucleotide Sequencing and Phylogenetic Analysis
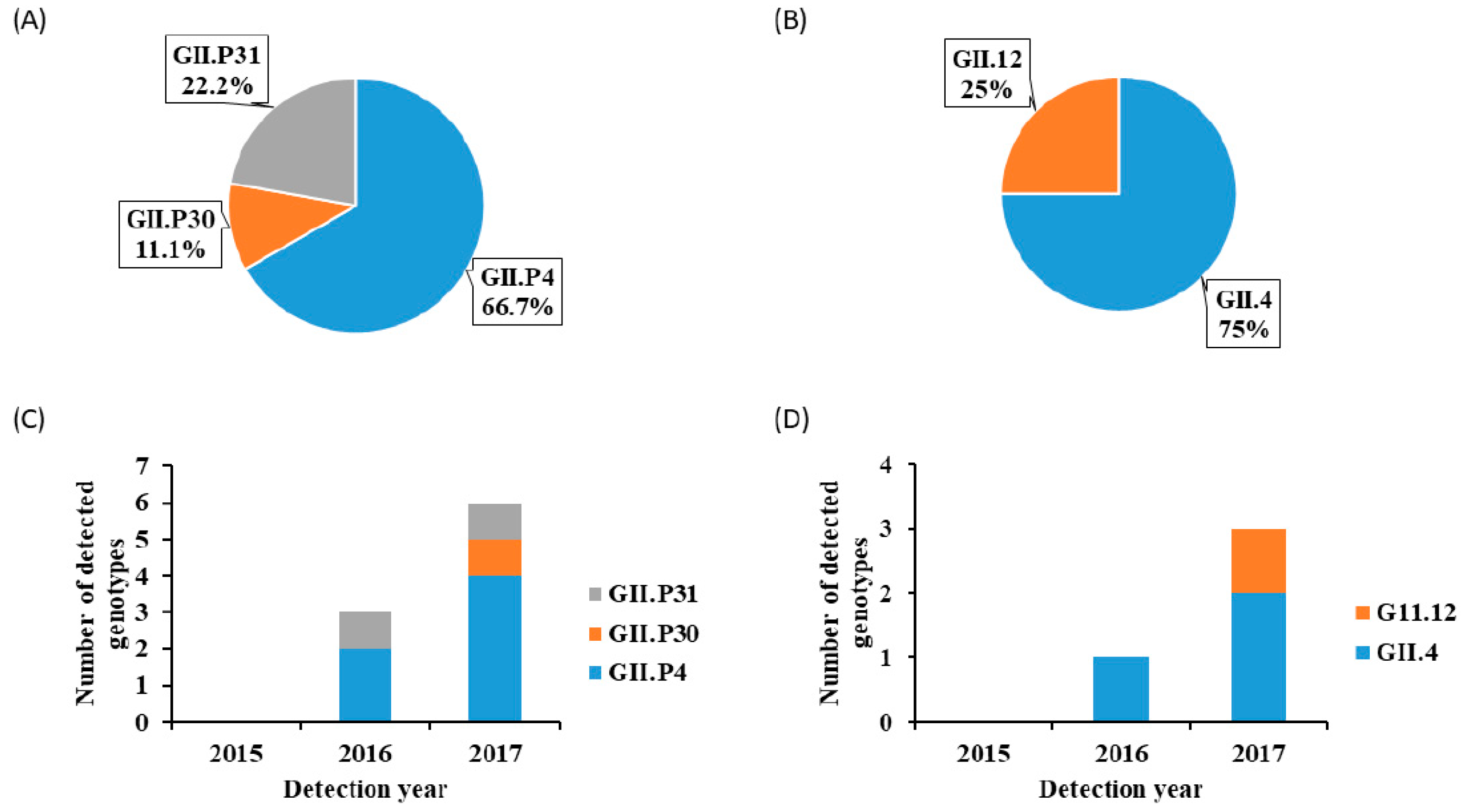
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Troeger, C.; Blacker, B.; Khalil, I.; Rao, P.; Cao, S.; Zimsen, S.; Albertson, S.B.; Stanaway, J.D.; Deshpande, A.; Abebe, Z.; et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1211–1228. [Google Scholar] [CrossRef]
- Bányai, K.; Estes, M.K.; Martella, V.; Parashar, U.D. Viral gastroenteritis. Lancet 2018, 392, 175–186. [Google Scholar] [CrossRef]
- Rota Global Introduction Status. Available online: http://preventrotavirus.org/vaccine-introduction/global-introduction-status/ (accessed on 15 May 2022).
- Ahmed, S.M.; Hall, A.J.; Robinson, A.E.; Verhoef, L.; Premkumar, P.; Parashar, U.D.; Koopmans, M.; Lopman, B.A. Global prevalence of norovirus in cases of gastroenteritis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 725–730. [Google Scholar] [CrossRef]
- Lopman, B.A.; Steele, D.; Kirkwood, C.D.; Parashar, U.D. The vast and varied global burden of norovirus: Prospects for prevention and control. PLoS Med. 2016, 13, e1001999. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, M.; Wang, K.; Estes, M.K. Sequence and Genomic Organization of Norwalk Virus. Virology 1993, 195, 51–61. [Google Scholar] [CrossRef]
- Thorne, L.G.; Goodfellow, I.G. Norovirus Gene Expression and Replication. J. Gen. Virol. 2014, 95, 278–291. [Google Scholar] [CrossRef]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef]
- Vinjé, J. Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef]
- Kroneman, A.; Vega, E.; Vennema, H.; Vinjé, J.; White, P.A.; Hansman, G.; Green, K.; Martella, V.; Katayama, K.; Koopmans, M. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 2013, 158, 2059–2068. [Google Scholar] [CrossRef]
- van Beek, J.; de Graaf, M.; Al-Hello, H.; Allen, D.J.; Ambert-Balay, K.; Botteldoorn, N.; Brytting, M.; Buesa, J.; Cabrerizo, M.; Chan, M.; et al. Molecular surveillance of norovirus, 2005–2016: An epidemiological analysis of data collected from the NoroNet network Lancet Infect. Dis. 2018, 18, 545–553. [Google Scholar]
- Chan, M.C.; Roy, S.; Bonifacio, J.; Zhang, L.-Y.; Chhabra, P.; Chan, J.C.; Celma, C.; Igoy, M.A.; Lau, S.-L.; Mohammad, K.N.; et al. Detection of norovirus variant GII. 4 Hong Kong in Asia and Europe, 2017–2019. Emerg. Infect. Dis. 2021, 27, 289–293. [Google Scholar] [CrossRef]
- Makhaola, K.; Moyo, S.; Lechiile, K.; Goldfarb, D.M.; Kebaabetswe, L.P. Genetic and epidemiological analysis of norovirus from children with gastroenteritis in Botswana, 2013–2015. BMC Infect. Dis. 2018, 18, 246–253. [Google Scholar] [CrossRef]
- Nordgren, J.; Svensson, L. Genetic susceptibility to human norovirus infection: An update. Viruses 2019, 11, 226. [Google Scholar] [CrossRef]
- Bull, R.A.; Hansman, G.S.; Clancy, L.E.; Tanaka, M.M.; Rawlinson, W.D.; White, P.A. Norovirus recombination in ORF1/ORF2 overlap. Emerg. Infect. Dis. 2005, 11, 1079–1085. [Google Scholar] [CrossRef]
- Cates, J.E.; Vinjé, J.; Parashar, U.; Hall, A.J. Recent advances in human norovirus research and implications for candidate vaccines. Expert Rev. Vaccines 2020, 19, 539–548. [Google Scholar] [CrossRef]
- Mans, J. Norovirus infections and disease in Lower-middle and Low-income countries, 1997–2018. Viruses 2019, 11, 341. [Google Scholar] [CrossRef]
- Afework, D.T.; Shumie, M.K.; Endalew, G.F.; Adugna, A.G.; Tarekegn, B.G. Pooled prevalence and genetic diversity of norovirus in Africa: A systematic review and meta-analysis. Virol. J. 2022, 19, 115. [Google Scholar] [CrossRef]
- Imade, P.E.; Eghafona, N.O. Viral agents of diarrhea in young children in two primary health centers in Edo State, Nigeria. Int. J. Microbiol. 2015, 2015, 685821. [Google Scholar] [CrossRef]
- Arowolo, K.O.; Ayolabi, C.I.; Lapinski, B.; Santos, J.S.; Raboni, S.M. Epidemiology of enteric viruses in children with gastroenteritis in Ogun State, Nigeria. J. Med. Virol. 2019, 91, 1022–1029. [Google Scholar] [CrossRef]
- Osazuwa, F.; Okojie, R.; Akinbo, F.O.; Johnson, W.; Grobler, H. Genetic diversity of norovirus among children under 5 years in South-South region of Nigeria. N. Z. J. Med. Lab. Sci. 2020, 74, 39–43. [Google Scholar]
- Japhet, M.O.; Adesina, O.A.; Famurewa, O.; Svensson, L.; Nordgren, J. Molecular epidemiology of rotavirus and norovirus in Ile-Ife, Nigeria: High prevalence of G12P[8] rotavirus strains and detection of a rare norovirus genotype. J. Med. Virol. 2012, 84, 1489–1496. [Google Scholar] [CrossRef]
- Japhet, M.O.; Famurewa, O.; Adesina, O.A.; Opaleye, O.O.; Wang, B.; Höhne, M.; Bock, C.T.; Marques, A.M.; Niendorf, S. Viral gastroenteritis among children of 0-5 years in Nigeria: Characterization of the first Nigerian aichivirus, recombinant noroviruses and detection of a zoonotic astrovirus. J. Clin. Virol. 2019, 111, 4–11. [Google Scholar] [CrossRef]
- Oyinloye, S.O.; Jatau, E.D.; Aminu, M.; Ella, E.E. A preliminary survey of norovirus and astrovirus antigen in diarrheic stools of children in Borno State, Nigeria. Glob. J. Med. Res. 2016, 16, 33–37. [Google Scholar]
- Ayolabi, C.I.; Ojo, D.A.; Armah, G.E.; Akpan, I.; Mafiana, C.F. Detection and partial characterization of norovirus among children with acute gastroenteritis in Lagos, Nigeria. Int. J. Med. Med. Sci. 2010, 2, 216–221. [Google Scholar]
- Arowolo, K.O.; Ayolabi, C.I.; Adeleye, I.A.; Lapinski, B.; Santos, J.S.; Raboni, S.M. Molecular epidemiology of astrovirus in children with gastroenteritis in southwestern Nigeria. Arch. Virol. 2020, 165, 2461–2469. [Google Scholar] [CrossRef]
- Vinje, J.; Koopmans, M.P. Molecular detection and epidemiology of small round structured viruses in outbreaks of gastroenteritis in the Netherlands. J. Infect. Dis. 1996, 174, 610–615. [Google Scholar] [CrossRef]
- Vinjé, J.; Hamidjaja, R.A.; Sobsey, M.D. Development and application of a capsid VP1 (region D) based reverse transcription PCR assay for genotyping of genogroup I and II noroviruses. J. Virol. Methods 2004, 116, 109–117. [Google Scholar] [CrossRef]
- Kroneman, A.; Vennema, H.; Deforche, K.; v d Avoort, H.; Peñaranda, S.; Oberste, M.S.; Vinjé, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.-T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.-S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef]
- Ouédraogo, N.; Kaplon, J.; Bonkoungou, I.J.; Traoré, A.S.; Pothier, P.; Barro, N.; Ambert-Balay, K. Prevalence and genetic diversity of enteric viruses in children with diarrhea in Ouagadougou, Burkina Faso. PLoS ONE 2016, 11, e0153652. [Google Scholar] [CrossRef]
- Mikounou Louya, V.; Vouvoungui, C.; Koukouikila-Koussounda, F.; Veas, F.; Kobawila, S.C.; Ntoumi, F. Molecular characterization of norovirus infection responsible for acute diarrhea in Congolese hospitalized children under five years old in Brazzaville, Republic of Congo. Int. J. Infect. Dis. 2019, 88, 41–48. [Google Scholar] [CrossRef]
- Mans, J.; Murray, T.Y.; Kiulia, N.M.; Mwenda, J.M.; Musoke, R.N.; Taylor, M.B. Human caliciviruses detected in HIV-seropositive children in Kenya. J. Med. Virol. 2014, 86, 75–81. [Google Scholar] [CrossRef]
- Siqueira, J.A.M.; Sousa Júnior, E.C.; Linhares, A.D.C.; Gabbay, Y.B. Molecular analysis of norovirus in specimens from children enrolled in a 1982-1986 study in Belém, Brazil: A community-based longitudinal study. J. Med. Virol. 2017, 89, 1539–1549. [Google Scholar] [CrossRef]
- Rossouw, E.; Brauer, M.; Meyer, P.; du Plessis, N.M.; Avenant, T.; Mans, J. Virus etiology, diversity and clinical characteristics in south african children hospitalised with gastroenteritis. Viruses 2021, 13, 215. [Google Scholar] [CrossRef]
- Hoa-Tran, T.N.; Nakagomi, O.; Dao, A.; Nguyen, A.T.; Agbemabiese, C.A.; Vu, H.M.; Nakagomi, T.; Thanh, N. Molecular epidemiology of noroviruses detected in Vietnamese children with acute gastroenteritis from 2012 to 2015. J. Med. Microbiol. 2017, 66, 34–45. [Google Scholar] [CrossRef]
- Biscaro, V.; Piccinelli, G.; Gargiulo, F.; Ianiro, G.; Caruso, A.; Caccuri, F.; De Francesco, M.A. Detection and molecular characterization of enteric viruses in children with acute gastroenteritis in Northern Italy. Infect. Genet. Evol. 2018, 60, 35–41. [Google Scholar] [CrossRef]
- Mans, J.; Murray, T.Y.; Nadan, S.; Netshikweta, R.; Page, N.A.; Taylor, M.B. Norovirus diversity in children with gastroenteritis in South Africa from 2009 to 2013: GII.4 variants and recombinant strains predominate. Epidemiol. Infect. 2016, 144, 907–916. [Google Scholar] [CrossRef]
- Kabue, J.P.; Meader, E.; Hunter, P.R.; Potgieter, N. Genetic characterisation of Norovirus strains in outpatient children from rural communities of Vhembe district/South Africa, 2014–2015. J. Clin. Virol. 2017, 94, 100–106. [Google Scholar] [CrossRef]
- Lu, L.; Zhong, H.; Xu, M.; Su, L.; Cao, L.; Jia, R.; Xu, J. Genetic diversity and epidemiology of Genogroup II noroviruses in children with acute sporadic gastroenteritis in Shanghai, China, 2012–2017. BMC Infect. Dis. 2019, 19, 736. [Google Scholar] [CrossRef]
- Hardy, M.E.; Kramer, S.F.; Treanor, J.J.; Estes, M.K. Human calicivirus genogroup II capsid sequence diversity revealed by analyses of the prototype Snow Mountain agent. Arch. Virol. 1997, 142, 1469–1479. [Google Scholar] [CrossRef]
- Hernandez, J.M.; Silva, L.D.; Junior, E.C.S.; Bandeira, R.S.; Rodrigues, E.A.M.; Lucena, M.S.S.; Costa, S.T.P.; Gabbay, Y.B. Molecular epidemiology and temporal evolution of norovirus associated with acute gastroenteritis in Amazonas state, Brazil. BMC Infect. Dis. 2018, 18, 147. [Google Scholar] [CrossRef]
- Cannon, J.L.; Bonifacio, J.; Bucardo, F.; Buesa, J.; Bruggink, L.; Chan, M.C.; Fumian, T.M.; Giri, S.; Gonzalez, M.D.; Hewitt, J.; et al. Global Trends in Norovirus Genotype Distribution among Children with Acute Gastroenteritis. Emerg. Infect. Dis. 2021, 27, 1438–1445. [Google Scholar] [CrossRef]
- Sisay, Z.; Djikeng, A.; Berhe, N.; Belay, G.; Gebreyes, W.; Abegaz, W.E.; Njahira, M.N.; Wang, Q.H.; Saif, L.J. Prevalence and molecular characterization of human noroviruses and sapoviruses in Ethiopia. Arch. Virol. 2016, 161, 2169–2182. [Google Scholar] [CrossRef]
- Bruggink, L.D.; Dunbar, N.L.; Marshall, J.A. Emergence of GII.e as a major ORF 1 norovirus genotype and its associated ORF 2 GII.4 variant forms. Infect. Genet. Evol. 2014, 22, 157–163. [Google Scholar] [CrossRef]
- Kõivumägi, K.; Geller, J.; Toompere, K.; Soeorg, H.; Kallas, E.; Jõgeda, E.L.; Huik, K.; Lutsar, I. Norovirus strains among children aged 0-18 years hospitalized with acute gastroenteritis in Estonia 2015–2016. J. Med. Virol. 2022, 94, 2632–2639. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arowolo, K.O.; Ayolabi, C.I.; Adeleye, I.A.; Lapinski, B.A.; Santos, J.S.; Raboni, S.M. Genetic Diversity of Norovirus in Children with Acute Gastroenteritis in Southwest Nigeria, 2015–2017. Viruses 2023, 15, 644. https://doi.org/10.3390/v15030644
Arowolo KO, Ayolabi CI, Adeleye IA, Lapinski BA, Santos JS, Raboni SM. Genetic Diversity of Norovirus in Children with Acute Gastroenteritis in Southwest Nigeria, 2015–2017. Viruses. 2023; 15(3):644. https://doi.org/10.3390/v15030644
Chicago/Turabian StyleArowolo, Kafayat O., Christianah I. Ayolabi, Isaac A. Adeleye, Bruna A. Lapinski, Jucelia S. Santos, and Sonia M. Raboni. 2023. "Genetic Diversity of Norovirus in Children with Acute Gastroenteritis in Southwest Nigeria, 2015–2017" Viruses 15, no. 3: 644. https://doi.org/10.3390/v15030644
APA StyleArowolo, K. O., Ayolabi, C. I., Adeleye, I. A., Lapinski, B. A., Santos, J. S., & Raboni, S. M. (2023). Genetic Diversity of Norovirus in Children with Acute Gastroenteritis in Southwest Nigeria, 2015–2017. Viruses, 15(3), 644. https://doi.org/10.3390/v15030644