
HuCoPIA: An Atlas of Human vs. SARS-CoV-2 Interactome and the Comparative Analysis with Other Coronaviridae Family Viruses



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
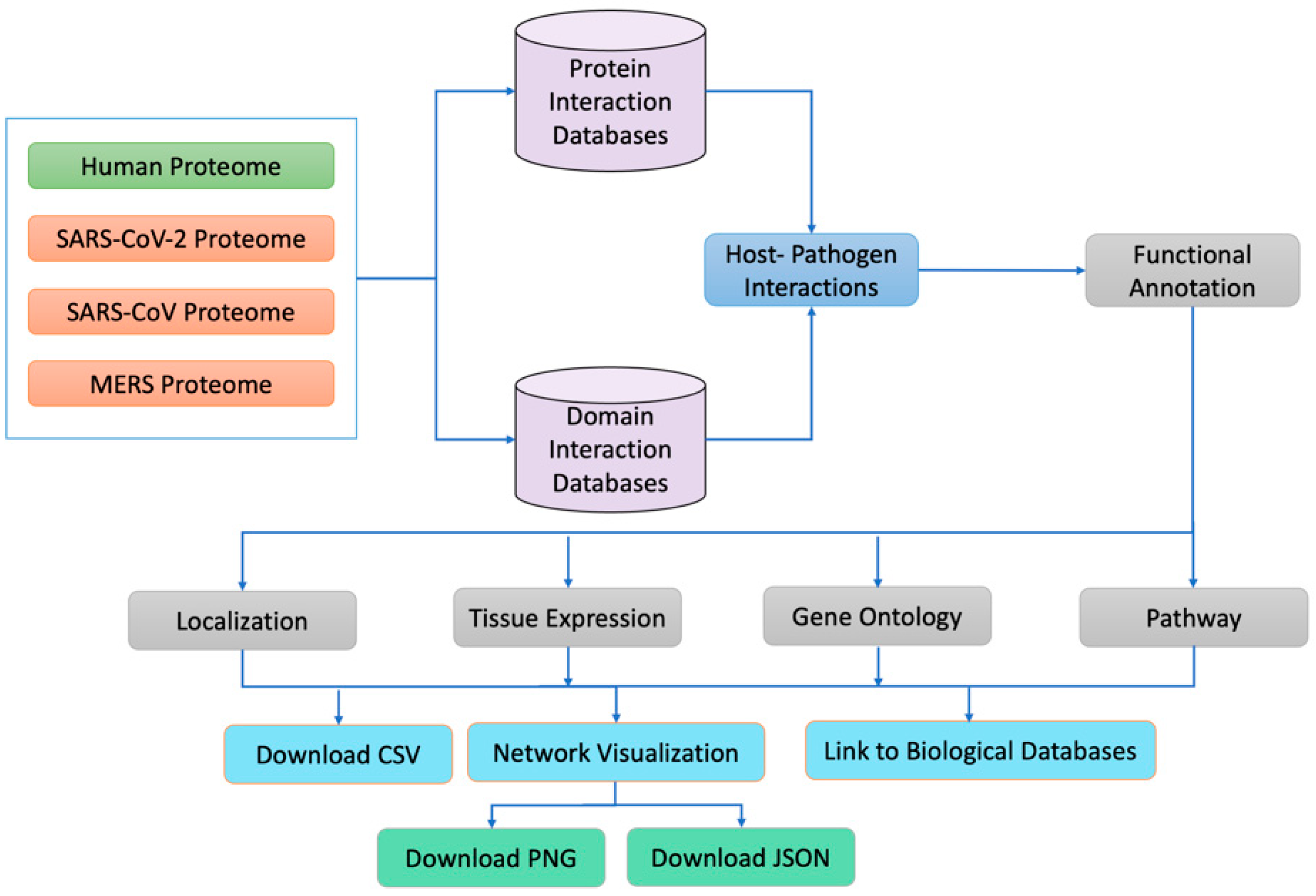
2. Material and Methods
2.1. Data Sources
2.2. Computational Prediction of Host Pathogen Interactions
2.3. Subcellular Localization of Human Proteins Involved in PPIs
2.4. Tissue Specificity of Human Proteins Involved in PPIs
2.5. Gene Ontology and KEGG Pathways Involved in PPIs
2.6. Implementation
3. Results
3.1. Interolog-Based Interaction Prediction
3.2. Domain-Based Interaction Prediction
3.3. Integration of Results from Two Approaches
3.4. HuCoPIA PPI’s Evaluation
4. Usage
5. Case Study
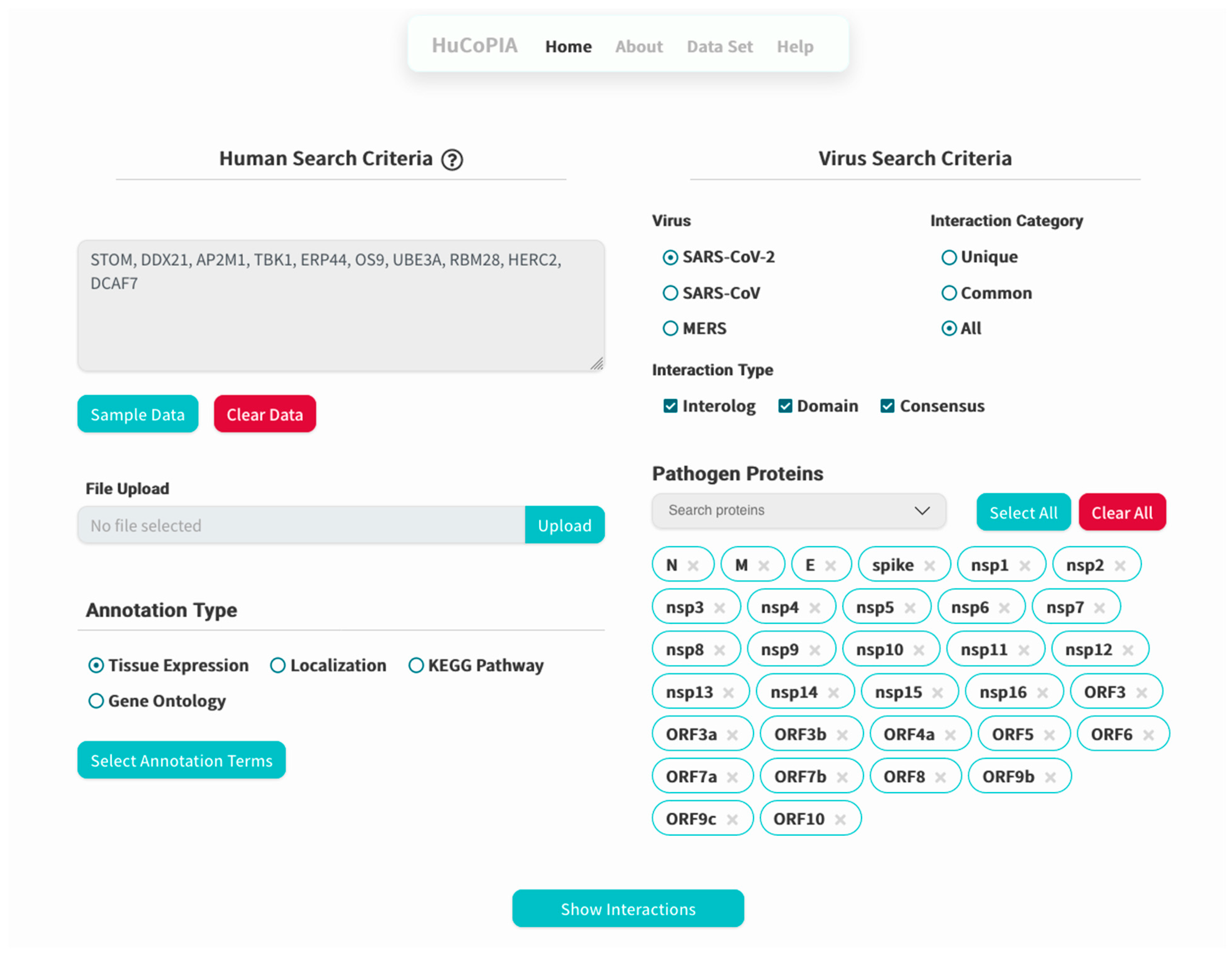
- Upload the genes text file on HuCoPIA (http://bioinfo.usu.edu/hucopia/, last accessed on 22 December 2022), select all virus proteins from the dropdown for each virus, and select tissue expression as the annotation type. Figure 2 shows the homepage of HuCoPIA with the search options. An intermediate results page will be shown after ‘show interactions’ page.
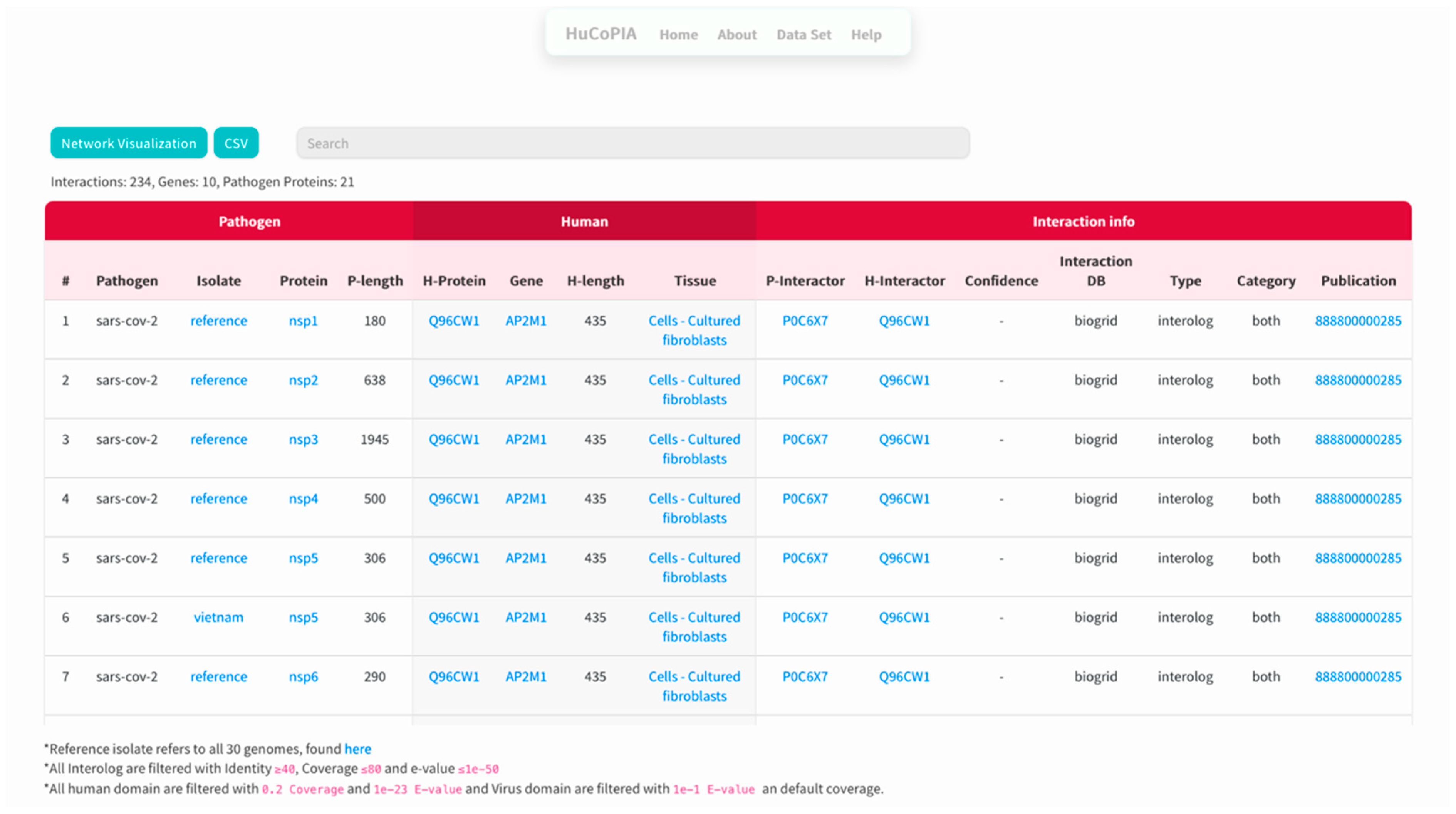
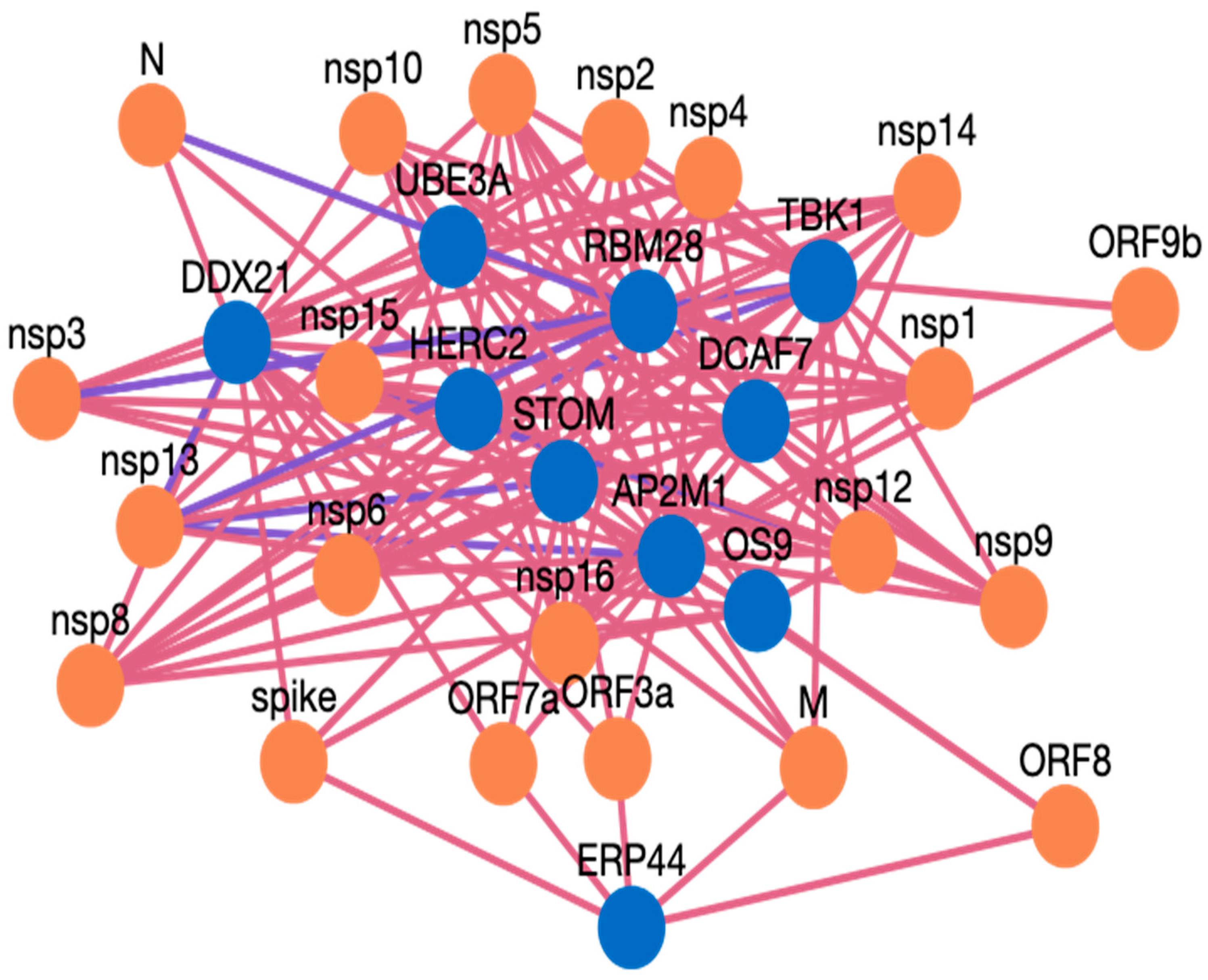
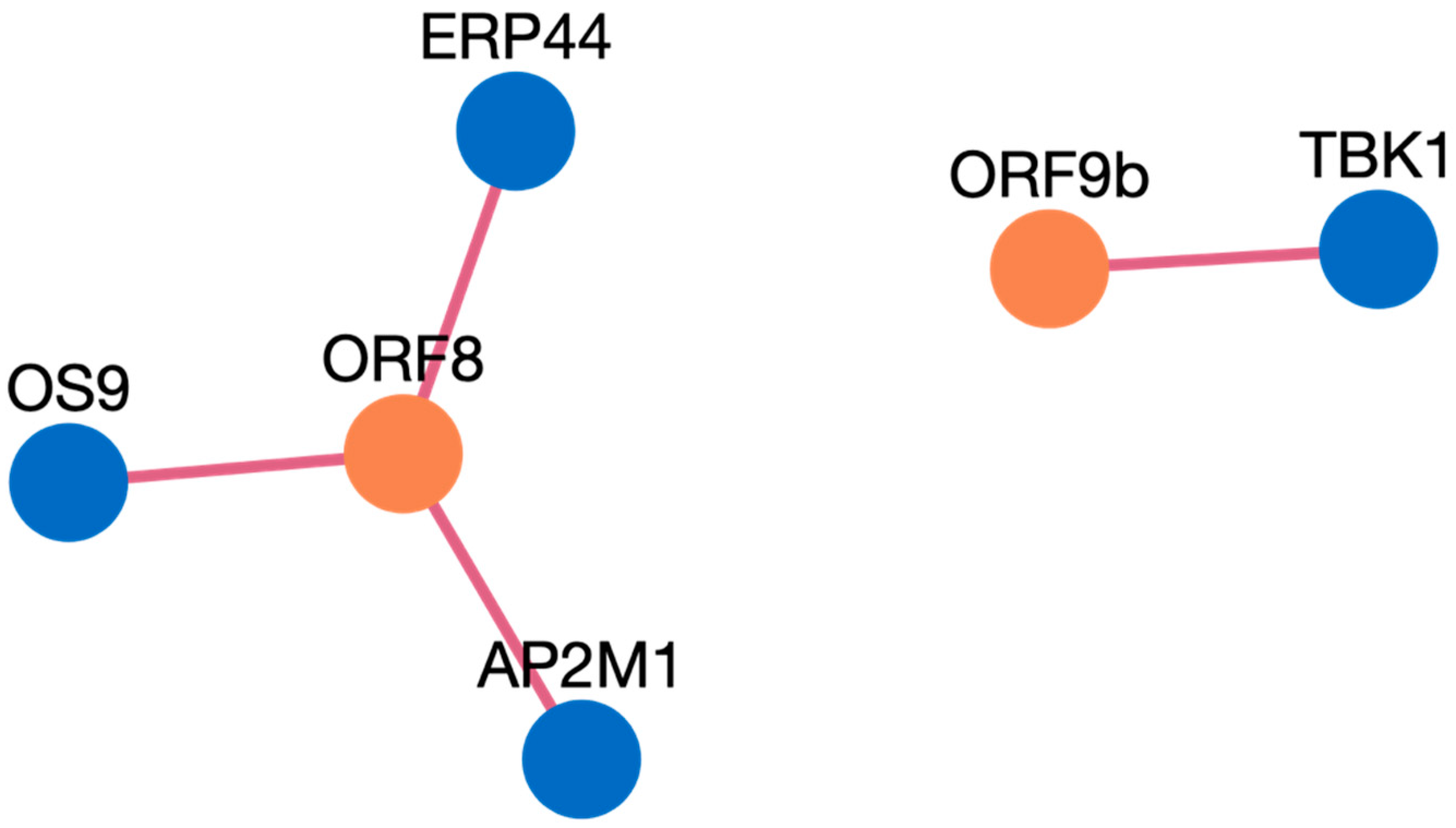
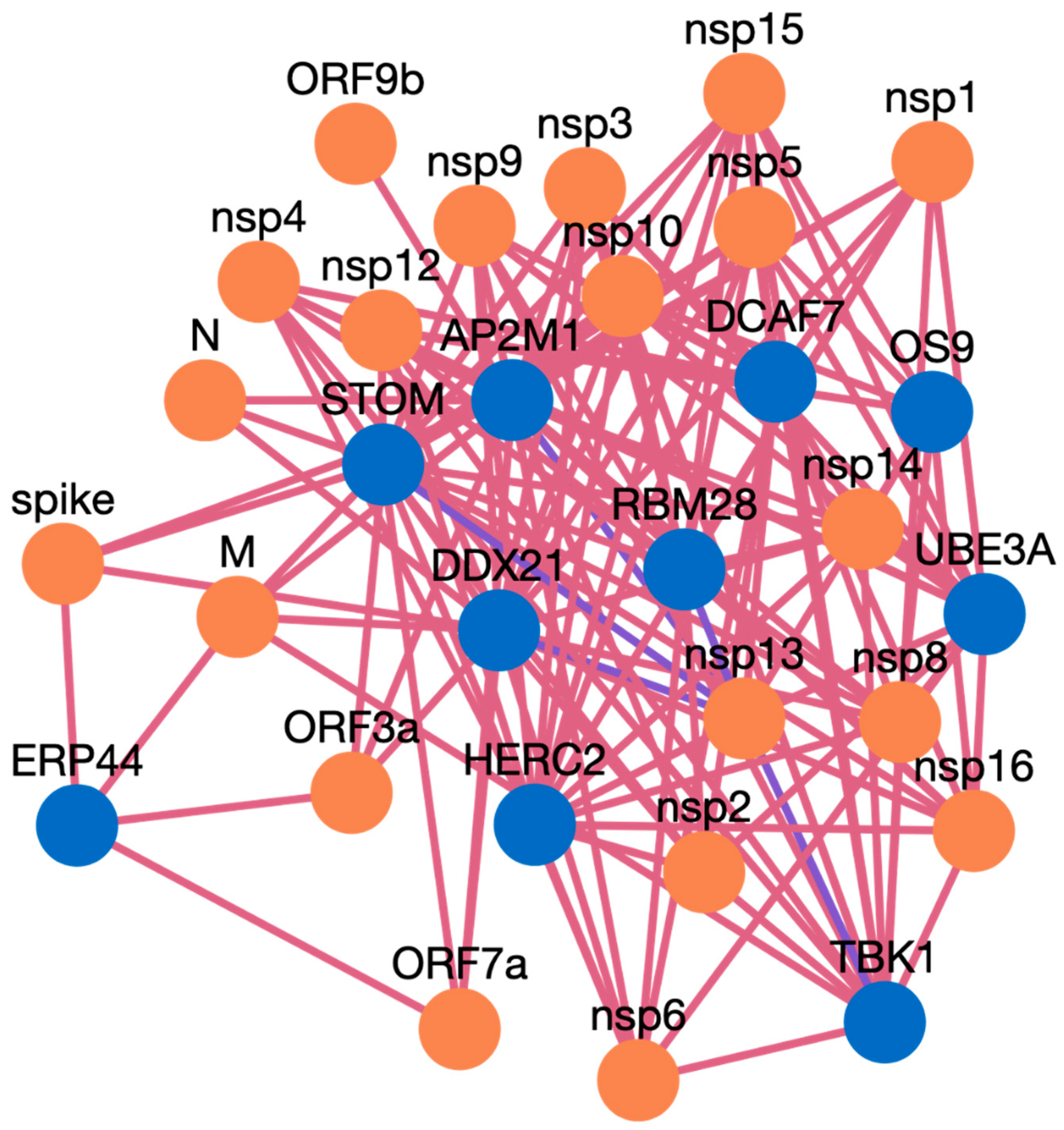
- The search results are displayed in a table. Information such as pathogen protein, pathogen isolate, pathogen protein length, human gene, human protein, human protein length, functional annotation, interaction source database, confidence, and PubMed links are available. Figure 3 depicts the results page. Further, a user can download the results as a csv file or visualize them in a network. Users can click on the proteins, or other links in blue, to check the detailed information. For the ten genes we submitted, there are 234 interactions involving 21 virus proteins. The results are presented in Supplementary Table S3.
Case Study Genes Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Tan, W.; Zhao, X.; Ma, X.; Wang, W.; Niu, P.; Xu, W.; Gao, G.F.; Wu, G.Z. A Novel Coronavirus Genome Identified in a Cluster of Pneumonia Cases—Wuhan, China 2019- 2020. China CDC Wkly. 2020, 2, 61–62. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.F. From “A”IV to “Z”IKV: Attacks from Emerging and Re-Emerging Pathogens. Cell 2018, 172, 1157–1159. [Google Scholar] [CrossRef]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.R.; Leibowitz, J.L. Coronavirus Pathogenesis. In Advances in Virus Research; Academic Press Inc.: Cambridge, MA, USA, 2011; Volume 81, pp. 85–164. [Google Scholar]
- Loaiza, C.D.; Duhan, N.; Lister, M.; Kaundal, R. In Silico Prediction of Host–Pathogen Protein Interactions in Melioidosis Pathogen Burkholderia Pseudomallei and Human Reveals Novel Virulence Factors and Their Targets. Brief. Bioinform. 2020, 22, bbz162. [Google Scholar] [CrossRef]
- Matthews, L.R.; Vaglio, P.; Reboul, J.; Ge, H.; Davis, B.P.; Garrels, J.; Vincent, S.; Vidal, M. Identification of Potential Interaction Networks Using Sequence-Based Searches for Conserved Protein-Protein Interactions or “Interologs”. Genome Res. 2001, 11, 2120–2126. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-K.; Zhang, Z.; Tan, S.-H. Integrative Approach for Computationally Inferring Protein Domain Interactions. Bioinformatics 2003, 19, 923–929. [Google Scholar] [CrossRef]
- Dyer, M.D.; Murali, T.M.; Sobral, B.W. Computational Prediction of Host-Pathogen Protein–Protein Interactions. Bioinformatics 2007, 23, i159–i166. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, L.; Guo, J.; Zhang, D.-Y.; Lin, K. Prediction of Yeast Protein-Protein Interaction Network: Insights from the Gene Ontology and Annotations. Nucleic Acids Res. 2006, 34, 2137–2150. [Google Scholar] [CrossRef]
- Davis, F.P.; Barkan, D.T.; Eswar, N.; McKerrow, J.H.; Sali, A. Host-Pathogen Protein Interactions Predicted by Comparative Modeling. Protein Sci. 2007, 16, 2585–2596. [Google Scholar] [CrossRef]
- Ogmen, U.; Keskin, O.; Aytuna, A.S.; Nussinov, R.; Gursoy, A. PRISM: Protein Interactions by Structural Matching. Nucleic Acids Res. 2005, 33, W331–W336. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-G.; He, F.; Zhang, Z.; Peng, Y.-L. Prediction of Protein–Protein Interactions between Ralstonia Solanacearum and Arabidopsis Thaliana. Amino Acids 2012, 42, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Syed Musthaq, S.; Kwang, J. Oral Vaccination of Baculovirus-Expressed VP28 Displays Enhanced Protection against White Spot Syndrome Virus in Penaeus Monodon. PLoS ONE 2011, 6, e26428. [Google Scholar] [CrossRef]
- Sahu, S.S.; Weirick, T.; Kaundal, R. Predicting Genome-Scale Arabidopsis-Pseudomonas Syringae Interactome Using Domain and Interolog-Based Approaches. BMC Bioinform. 2014, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, B.A.; Panchenko, A.R. Deciphering Protein–Protein Interactions. Part II. Computational Methods to Predict Protein and Domain Interaction Partners. PLoS Comput. Biol. 2007, 3, e43. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Rezaei, J.; Hugo, W.; Gao, S.; Jin, J.; Fan, M.; Yong, C.-H.; Wozniak, M.; Wong, L. Stringent DDI-Based Prediction of H. Sapiens-M. Tuberculosis H37Rv Protein-Protein Interactions. BMC Syst. Biol. 2013, 7, S6. [Google Scholar] [CrossRef]
- Ghosh, N.; Saha, I.; Sharma, N. Interactome of Human and SARS-CoV-2 Proteins to Identify Human Hub Proteins Associated with Comorbidities. Comput. Biol. Med. 2021, 138, 104889. [Google Scholar] [CrossRef]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative Host-Coronavirus Protein Interaction Networks Reveal Pan-Viral Disease Mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, Y.; Gupta, S.; Paramo, M.I.; Hou, Y.; Mao, C.; Luo, Y.; Judd, J.; Wierbowski, S.; Bertolotti, M.; et al. A Comprehensive SARS-CoV-2–Human Protein–Protein Interactome Reveals COVID-19 Pathobiology and Potential Host Therapeutic Targets. Nat. Biotechnol. 2022, 41, 128–139. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Jian, X.; Zhang, Y.; Zhao, J.; Zhao, Z.; Lu, M.; Xie, L. CoV2-TCR: A Web Server for Screening TCR CDR3 from TCR Immune Repertoire of COVID-19 Patients and Their Recognized SARS-CoV-2 Epitopes. Comput. Struct. Biotechnol. J. 2023. [Google Scholar] [CrossRef]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-Infected Host Cells Reveals Therapy Targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel Proteomics Reveals Host Perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Lareau, C.A.; Keshishian, H.; Ganskih, S.; Schneider, C.; Hennig, T.; Melanson, R.; Werner, S.; Wei, Y.; Zimmer, M.; et al. The SARS-CoV-2 RNA–Protein Interactome in Infected Human Cells. Nat. Microbiol. 2021, 6, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y.; et al. Virus–Host Interactome and Proteomic Survey Reveal Potential Virulence Factors Influencing SARS-CoV-2 Pathogenesis. Med 2021, 2, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Lasso, G.; Mayer, S.V.; Winkelmann, E.R.; Chu, T.; Elliot, O.; Patino-Galindo, J.A.; Park, K.; Rabadan, R.; Honig, B.; Shapira, S.D. A Structure-Informed Atlas of Human-Virus Interactions. Cell 2019, 178, 1526–1541.e16. [Google Scholar] [CrossRef]
- Ammari, M.G.; Gresham, C.R.; McCarthy, F.M.; Nanduri, B. HPIDB 2.0: A Curated Database for Host-Pathogen Interactions. Database 2016, 2016, baw103. [Google Scholar] [CrossRef]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct Molecular Interaction Database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef]
- Xenarios, I.; Rice, D.W.; Salwinski, L.; Baron, M.K.; Marcotte, E.M.; Eisenberg, D. DIP: The Database of Interacting Proteins. Nucleic Acids Res. 2000, 28, 289–291. [Google Scholar] [CrossRef]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the Molecular Interaction Database: 2012 Update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID Interaction Database: 2019 Update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [PubMed]
- Ako-Adjei, D.; Fu, W.; Wallin, C.; Katz, K.S.; Song, G.; Darji, D.; Brister, J.R.; Ptak, R.G.; Pruitt, K.D. HIV-1, Human Interaction Database: Current Status and New Features. Nucleic Acids Res. 2015, 43, D566–D570. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam Protein Families Database. Nucleic Acids Res. 2009, 38, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Min, B.; Yi, G.S. IDDI: Integrated Domain-Domain Interaction and Protein Interaction Analysis System. Proteome Sci. 2012, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mosca, R.; Céol, A.; Stein, A.; Olivella, R.; Aloy, P. 3did: A Catalog of Domain-Based Interactions of Known Three-Dimensional Structure. Nucleic Acids Res. 2014, 42, D374. [Google Scholar] [CrossRef] [PubMed]
- Raghavachari, B.; Tasneem, A.; Przytycka, T.M.; Jothi, R. DOMINE: A Database of Protein Domain Interactions. Nucleic Acids Res. 2008, 36, D656. [Google Scholar] [CrossRef]
- Hung, M.C.; Link, W. Protein Localization in Disease and Therapy. J. Cell Sci. 2011, 124, 3381–3392. [Google Scholar] [CrossRef]
- Aguet, F.; Brown, A.A.; Castel, S.E.; Davis, J.R.; He, Y.; Jo, B.; Mohammadi, P.; Park, Y.S.; Parsana, P.; Segrè, A.V.; et al. Genetic Effects on Gene Expression across Human Tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Amirbakhtiar, N.; Ismaili, A.; Ghaffari, M.R.; Firouzabadi, F.N.; Shobbar, Z.S.; Fu, Z.Q.; Guo, M.; Jeong, B.R.; Tian, F.; Elthon, T.E.; et al. Plant ABC Transporters. Front. Plant Sci. 2017, 9, 1–15. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y.; Benjamini, Y.H.Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multple Testing. J. R. Stat. Society. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Bose, T.; Venkatesh, K.V.; Mande, S.S. Computational Analysis of Host–Pathogen Protein Interactions between Humans and Different Strains of Enterohemorrhagic Escherichia Coli. Front. Cell. Infect. Microbiol. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Blohm, P.; Frishman, G.; Smialowski, P.; Goebels, F.; Wachinger, B.; Ruepp, A.; Frishman, D. Negatome 2.0: A Database of Non-Interacting Proteins Derived by Literature Mining, Manual Annotation and Protein Structure Analysis. Nucleic Acids Res. 2014, 42, D396–D400. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chu, H.; Huang, J.; Zhao, X.; Ye, Z.W.; Lai, P.M.; Wen, L.; Cai, J.P.; Mo, Y.; Cao, J.; et al. Viruses Harness YxxØ Motif to Interact with Host AP2M1 for Replication: A Vulnerable Broad-Spectrum Antiviral Target. Sci. Adv. 2020, 6, 7910–7938. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.G.; Tang, D.J.; Hua, Z.; Wang, Z.; An, J. Sunitinib Reduces the Infection of SARS-CoV, MERS-CoV and SARS-CoV-2 Partially by Inhibiting AP2M1 Phosphorylation. Cell Discov. 2020, 6, 1–5. [Google Scholar] [CrossRef]
- Ariumi, Y. Host Cellular RNA Helicases Regulate SARS-CoV-2 Infection. J. Virol. 2022, 96, e00002-22. [Google Scholar] [CrossRef]
- Schmidt, A.; Peters, S.; Knaus, A.; Sabir, H.; Hamsen, F.; Maj, C.; Fazaal, J.; Sivalingam, S.; Savchenko, O.; Mantri, A.; et al. TBK1 and TNFRSF13B Mutations and an Autoinflammatory Disease in a Child with Lethal COVID-19. NPJ Genom. Med. 2021, 6, 1–5. [Google Scholar] [CrossRef]
- Sureda, A.; Alizadeh, J.; Nabavi, S.F.; Berindan-Neagoe, I.; Cismaru, C.A.; Jeandet, P.; Łos, M.J.; Clementi, E.; Nabavi, S.M.; Ghavami, S. Endoplasmic Reticulum as a Potential Therapeutic Target for Covid-19 Infection Management? Eur. J. Pharmacol. 2020, 882, 173288. [Google Scholar] [CrossRef]
- Zheng, X.; Sun, Z.; Yu, L.; Shi, D.; Zhu, M.; Yao, H.; Li, L. Interactome Analysis of the Nucleocapsid Protein of SARS-CoV-2 Virus. Pathogens 2021, 10, 1155. [Google Scholar] [CrossRef]
- Noack, J.; Bernasconi, R.; Molinari, M. How Viruses Hijack the ERAD Tuning Machinery. J. Virol. 2014, 88, 10272. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, Y.G.; Zhang, H. Endomembrane Remodeling in SARS-CoV-2 Infection. Cell Insight 2022, 1, 100031. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Mehra, R.; Kallianpur, A.; Culver, D.A.; Gack, M.U.; Farha, S.; Zein, J.; Comhair, S.; et al. A Network Medicine Approach to Investigation and Population-Based Validation of Disease Manifestations and Drug Repurposing for COVID-19. PLoS Biol. 2020, 18, e3000970. [Google Scholar] [CrossRef] [PubMed]
- Achom, A.; Das, R.; Pakray, P. An Improved Fuzzy Based GWO Algorithm for Predicting the Potential Host Receptor of COVID-19 Infection. Comput. Biol. Med. 2022, 151, 106050. [Google Scholar] [CrossRef]
- Pinto, S.M.; Subbannayya, Y.; Kim, H.; Hagen, L.; Górna, M.W.; Nieminen, A.I.; Bjørås, M.; Espevik, T.; Kainov, D.; Kandasamy, R.K. Multi-OMICs Landscape of SARS-CoV-2-Induced Host Responses in Human Lung Epithelial Cells. iScience 2023, 26, 105895. [Google Scholar] [CrossRef]
- Thirumal Kumar, D.; Shree Devi, M.S.; Udhaya Kumar, S.; Sherlin, A.; Mathew, A.; Lakshmipriya, M.; Sathiyarajeswaran, P.; Gnanasambandan, R.; Siva, R.; Magesh, R.; et al. Understanding the Activating Mechanism of the Immune System against COVID-19 by Traditional Indian Medicine: Network Pharmacology Approach. Adv. Protein Chem. Struct. Biol. 2022, 129, 275. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duhan, N.; Kaundal, R. HuCoPIA: An Atlas of Human vs. SARS-CoV-2 Interactome and the Comparative Analysis with Other Coronaviridae Family Viruses. Viruses 2023, 15, 492. https://doi.org/10.3390/v15020492
Duhan N, Kaundal R. HuCoPIA: An Atlas of Human vs. SARS-CoV-2 Interactome and the Comparative Analysis with Other Coronaviridae Family Viruses. Viruses. 2023; 15(2):492. https://doi.org/10.3390/v15020492
Chicago/Turabian StyleDuhan, Naveen, and Rakesh Kaundal. 2023. "HuCoPIA: An Atlas of Human vs. SARS-CoV-2 Interactome and the Comparative Analysis with Other Coronaviridae Family Viruses" Viruses 15, no. 2: 492. https://doi.org/10.3390/v15020492
APA StyleDuhan, N., & Kaundal, R. (2023). HuCoPIA: An Atlas of Human vs. SARS-CoV-2 Interactome and the Comparative Analysis with Other Coronaviridae Family Viruses. Viruses, 15(2), 492. https://doi.org/10.3390/v15020492