
Phylogeography as a Proxy for Population Connectivity for Spatial Modeling of Foot-and-Mouth Disease Outbreaks in Vietnam

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Population Description
2.2. Data Overview
2.3. Sequence Acquisition
2.4. Phylodynamic Analyses
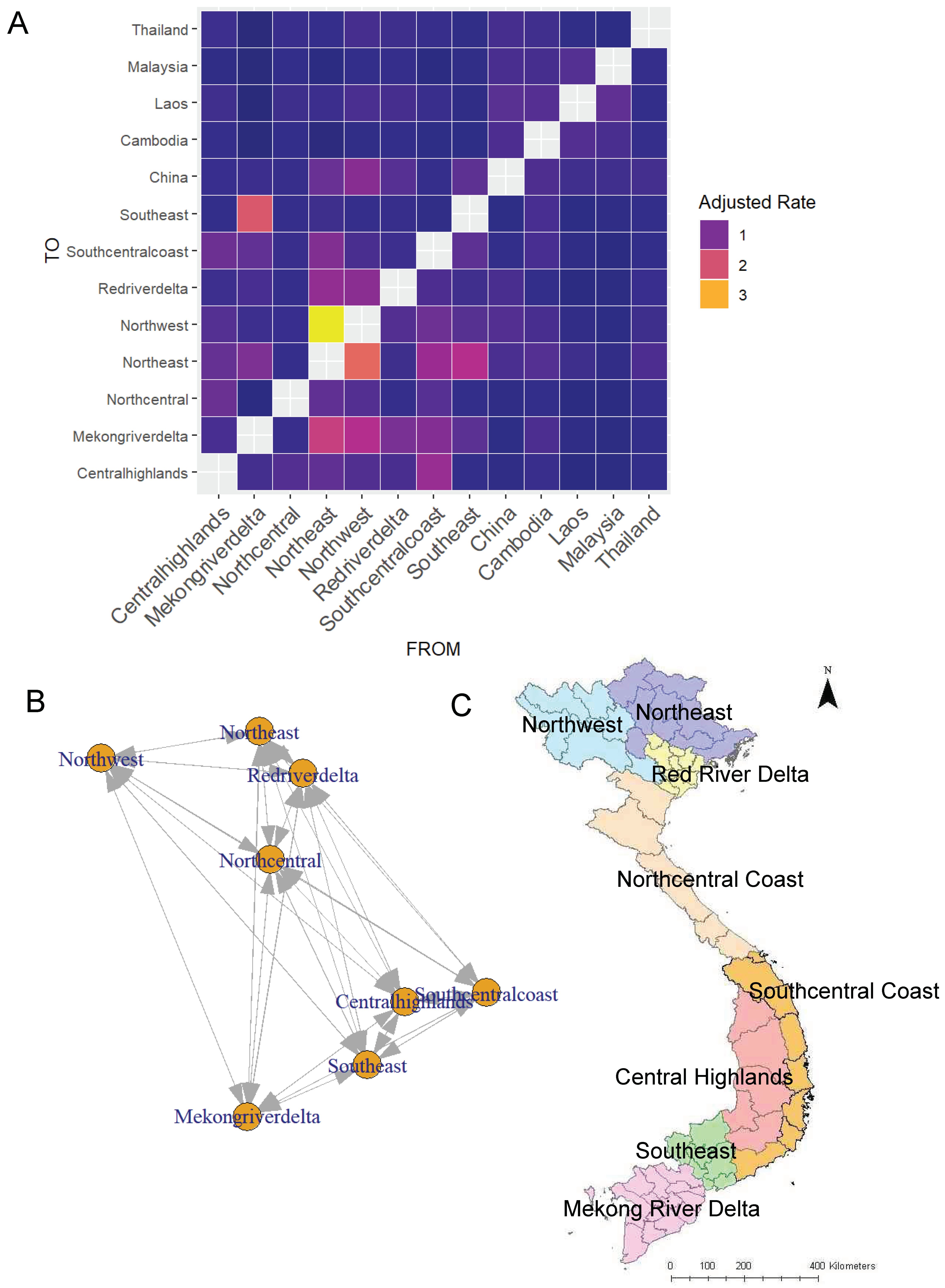
2.4.1. Calculation of Adjusted Transition Rates between Agricultural Zones
2.4.2. Analysis of Interspecies Transmission
2.5. Bayesian Space–Time Risk Models
2.5.1. Incorporation of Spatial Risk Factors as Fixed Effects
2.5.2. Prior Sensitivity Analysis and Evaluation of Model Fit
2.5.3. Software for Space–Time Regression
3. Results
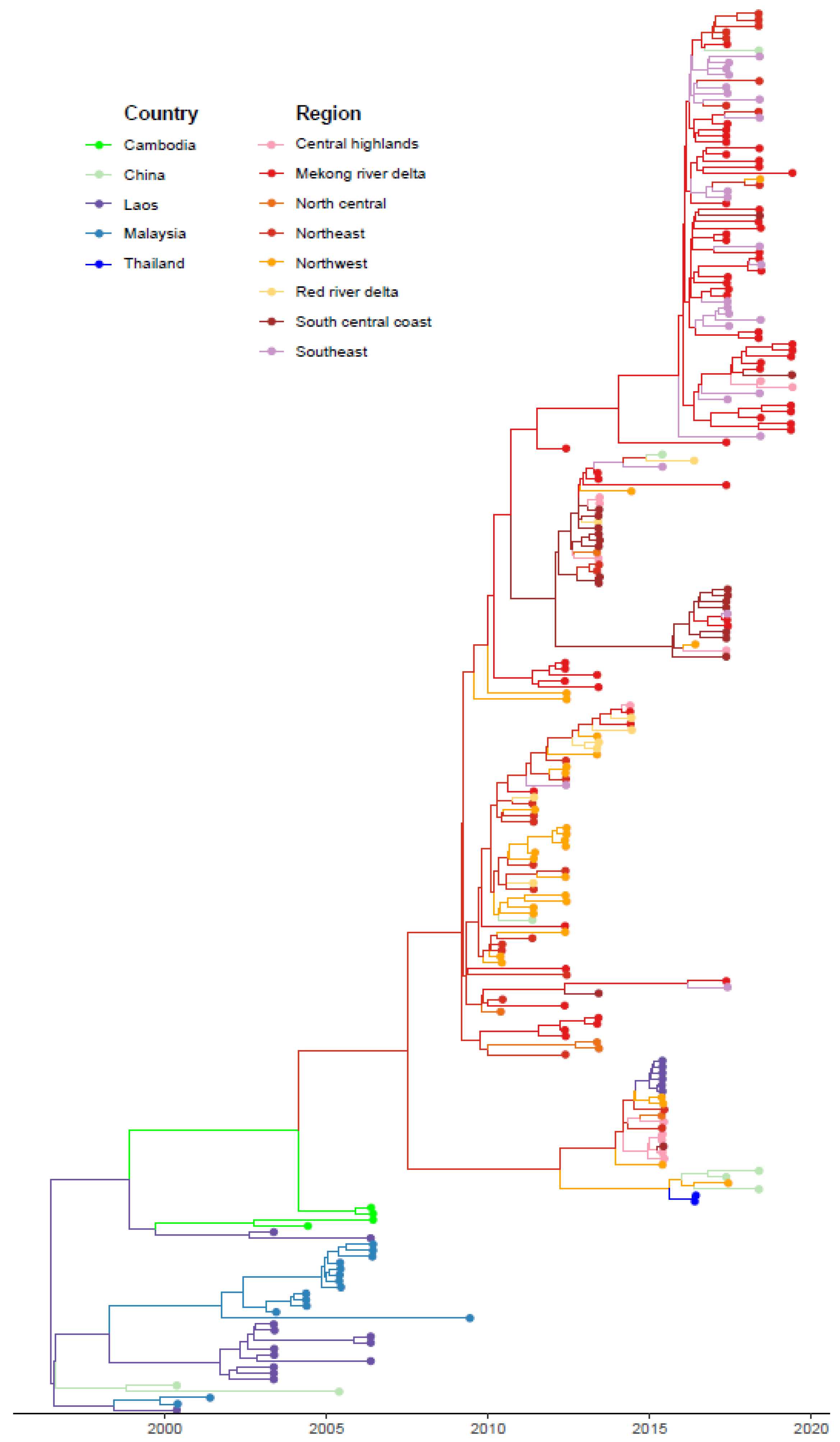
3.1. Phylogeographic Analyses
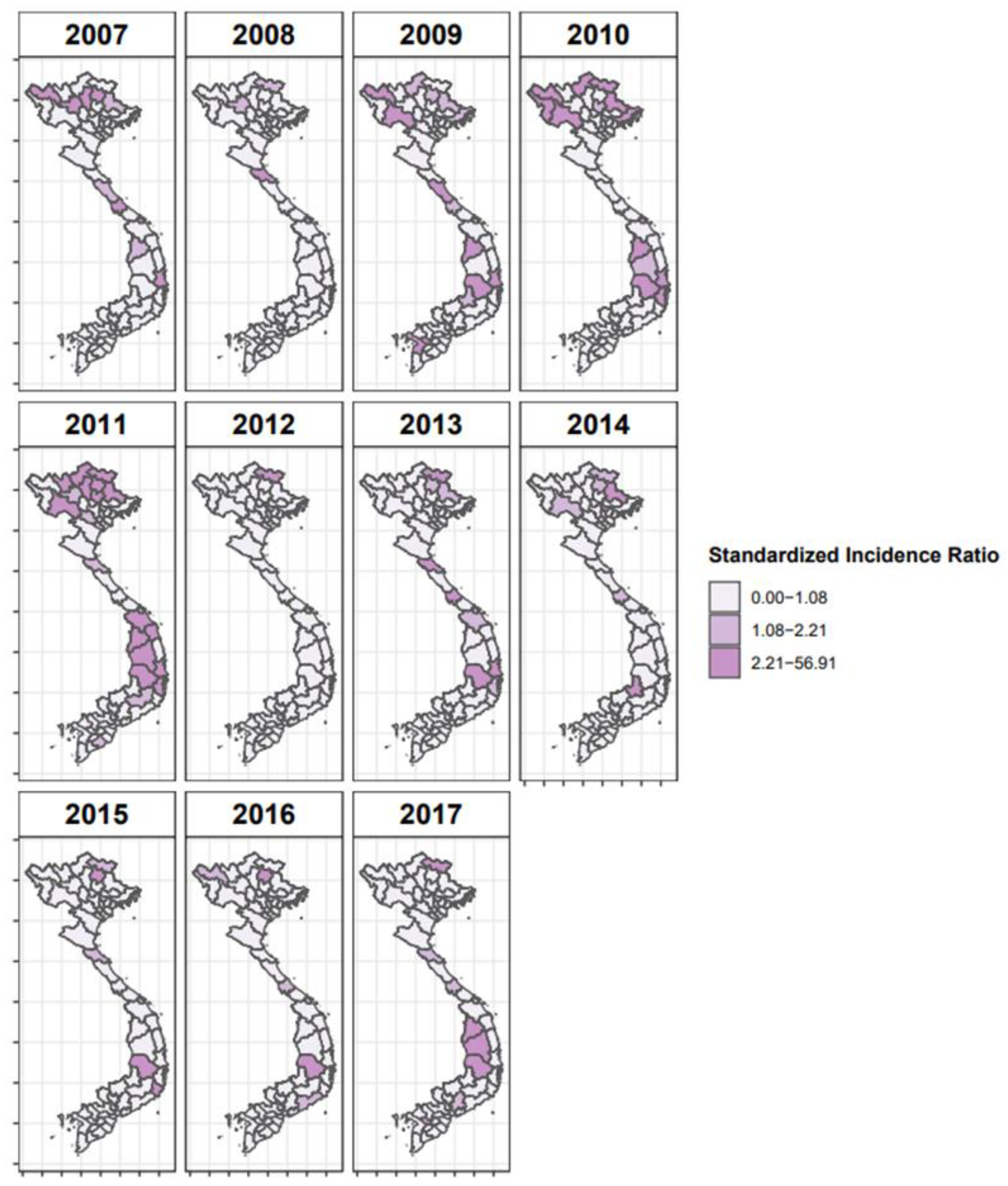
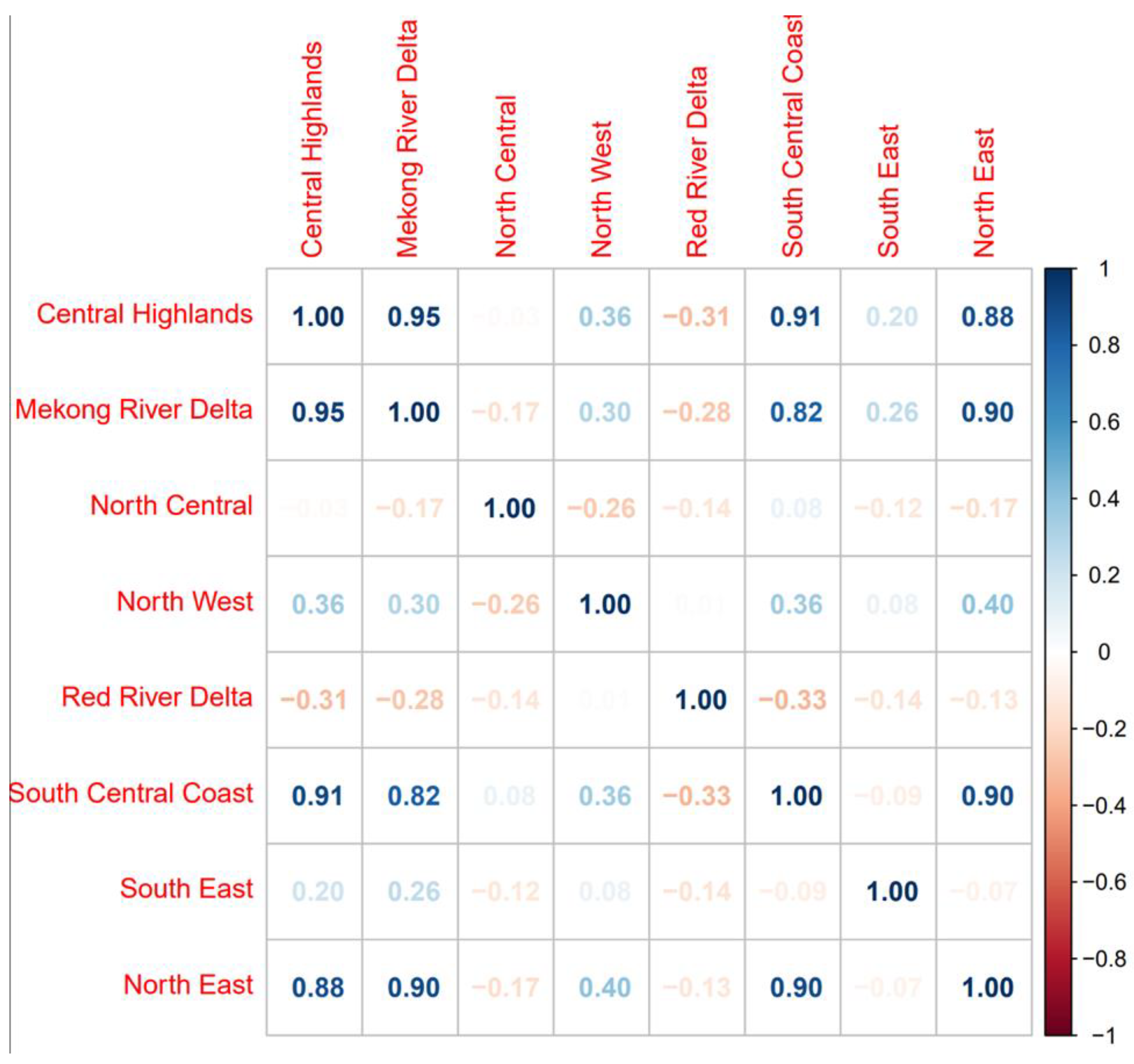
3.2. Space–Time Risk Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berliner, L.M. Hierarchical Bayesian Time Series Models. In Maximum Entropy and Bayesian Methods; Hanson, K.M., Silver, R.N., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 1996; pp. 15–22. [Google Scholar]
- Lawson, A.B. Bayesian Latent Modeling of Spatio-Temporal Variation in Small-Area Health Data. WIREs Comput. Stat. 2018, 10, e1441. [Google Scholar] [CrossRef]
- Machado, G.; Korennoy, F.; Alvarez, J.; Picasso-Risso, C.; Perez, A.; VanderWaal, K. Mapping Changes in the Spatiotemporal Distribution of Lumpy Skin Disease Virus. bioRxiv 2019, 2, 531343. [Google Scholar] [CrossRef] [PubMed]
- Blangiardo, M.; Cameletti, M. Spatial and Spatio-Temporal Bayesian Models with R—INLA; John Wiley & Sons: Hoboken, NJ, USA, 2015; ISBN 978-1-118-32655-8. [Google Scholar]
- Coly, S.; Garrido, M.; Abrial, D.; Yao, A.-F. Bayesian Hierarchical Models for Disease Mapping Applied to Contagious Pathologies. PLoS ONE 2021, 16, e0222898. [Google Scholar] [CrossRef] [PubMed]
- Fountain-Jones, N.M.; Kraberger, S.; Gagne, R.B.; Trumbo, D.R.; Salerno, P.E.; Chris Funk, W.; Crooks, K.; Biek, R.; Alldredge, M.; Logan, K.; et al. Host relatedness and landscape connectivity shape pathogen spread in the puma, a large secretive carnivore. Commun. Biol. 2021, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Dellicour, S.; Rose, R.; Faria, N.R.; Vieira, L.F.P.; Bourhy, H.; Gilbert, M.; Lemey, P.; Pybus, O.G. Using Viral Gene Sequences to Compare and Explain the Heterogeneous Spatial Dynamics of Virus Epidemics. Mol. Biol. Evol. 2017, 34, 2563–2571. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho Ferreira, H.C.; Pauszek, S.J.; Ludi, A.; Huston, C.L.; Pacheco, J.M.; Le, V.T.; Nguyen, P.T.; Bui, H.H.; Nguyen, T.D.; Nguyen, T.; et al. An Integrative Analysis of Foot-and-Mouth Disease Virus Carriers in Vietnam Achieved through Targeted Surveillance and Molecular Epidemiology. Transbound. Emerg. Dis. 2017, 64, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Le, V.P.; Vu, T.T.H.; Duong, H.-Q.; Than, V.T.; Song, D. Evolutionary Phylodynamics of Foot-and-Mouth Disease Virus Serotypes O and A Circulating in Vietnam. BMC Vet. Res. 2016, 12, 269. [Google Scholar] [CrossRef]
- Paton, D.J.; Gubbins, S.; King, D.P. Understanding the Transmission of Foot-and-Mouth Disease Virus at Different Scales. Curr. Opin. Virol. 2018, 28, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Rweyemamu, M.; Roeder, P.; Mackay, D.; Sumption, K.; Brownlie, J.; Leforban, Y.; Valarcher, J.-F.; Knowles, N.J.; Saraiva, V. Epidemiological Patterns of Foot-and-Mouth Disease Worldwide. Transbound. Emerg. Dis. 2008, 55, 57–72. [Google Scholar] [CrossRef]
- Stenfeldt, C.; Pacheco, J.M.; Smoliga, G.R.; Bishop, E.; Pauszek, S.J.; Hartwig, E.J.; Rodriguez, L.L.; Arzt, J. Detection of Foot-and-Mouth Disease Virus RNA and Capsid Protein in Lymphoid Tissues of Convalescent Pigs Does Not Indicate Existence of a Carrier State. Transbound. Emerg. Dis. 2016, 63, 152–164. [Google Scholar] [CrossRef]
- Bachanek-Bankowska, K.; Di Nardo, A.; Wadsworth, J.; Mioulet, V.; Pezzoni, G.; Grazioli, S.; Brocchi, E.; Kafle, S.C.; Hettiarachchi, R.; Kumarawadu, P.L.; et al. Reconstructing the Evolutionary History of Pandemic Foot-and-Mouth Disease Viruses: The Impact of Recombination within the Emerging O/ME-SA/Ind-2001 Lineage. Sci. Rep. 2018, 8, 14693. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.T.; Long, N.T.; Brito, B.; Stenfeldt, C.; Phuong, N.T.; Hoang, B.H.; Pauszek, S.J.; Hartwig, E.J.; Smoliga, G.R.; Vu, P.P.; et al. First Detection of Foot-and-Mouth Disease Virus O/Ind-2001d in Vietnam. PLoS ONE 2017, 12, e0177361. [Google Scholar] [CrossRef] [PubMed]
- Brito, B.; Pauszek, S.J.; Eschbaumer, M.; Stenfeldt, C.; de Carvalho Ferreira, H.C.; Vu, L.T.; Phuong, N.T.; Hoang, B.H.; Tho, N.D.; Dong, P.V.; et al. Phylodynamics of Foot-and-Mouth Disease Virus O/PanAsia in Vietnam 2010–2014. Vet. Res. 2017, 48, 24. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Knowles, N.J.; Wadsworth, J.; Haydon, D.T.; King, D.P. Phylodynamic Reconstruction of O CATHAY Topotype Foot-and-Mouth Disease Virus Epidemics in the Philippines. Vet. Res. 2014, 45, 90. [Google Scholar] [CrossRef]
- Polly, P.; Bourgeois Lüthi, N.; Huachun, L.; Naing Oo, K.; Phonvisay, A.; Premashthira, S.; Abila, R.; Widders, P.; Kukreja, K.; Miller, C. Movement Pathways and Market Chains of Large Ruminants in the Greater Mekong Sub-Region; World Organisation for Animal Health: Paris, France, 2015. [Google Scholar]
- Lee, H.S.; Pham, T.L.; Wieland, B. Temporal Patterns and Space-Time Cluster Analysis of Foot-and-Mouth Disease (FMD) Cases from 2007 to 2017 in Vietnam. Transbound. Emerg. Dis. 2020, 67, 584–591. [Google Scholar] [CrossRef]
- Pham-Thanh, L.; Magnusson, U.; Can-Xuan, M.; Nguyen-Viet, H.; Lundkvist, Å.; Lindahl, J. Livestock Development in Hanoi City, Vietnam—Challenges and Policies. Front. Vet. Sci. 2020, 7, 566. [Google Scholar] [CrossRef]
- Dinh, T.X. An Overview of Agricultural Pollution in Vietnam: The Livestock Sector; World Bank: Washington, DC, USA, 2007. [Google Scholar]
- Baudon, E.; Fournié, G.; Hiep, D.T.; Pham, T.T.; Duboz, R.; Gély, M.; Peiris, M.; Cowling, B.J.; Ton, V.D.; Peyre, M. Analysis of Swine Movements in a Province in Northern Vietnam and Application in the Design of Surveillance Strategies for Infectious Diseases. Transbound. Emerg. Dis. 2017, 64, 411–424. [Google Scholar] [CrossRef]
- Van Diep, N.; Ngoc, T.T.B.; Hoa, L.Q.; Nga, B.T.T.; Kang, B.; Oh, J.; Lan, N.T.; Le, V.P. O/SEA/Mya-98 Lineage Foot-and-Mouth Disease Virus Was Responsible for an Extensive Epidemic That Occurred in Late 2018 in Vietnam. Arch. Virol. 2020, 165, 2487–2493. [Google Scholar] [CrossRef] [PubMed]
- Knowles, N.J.; Samuel, A.R.; Davies, P.R.; Midgley, R.J.; Valarcher, J.-F. Pandemic Strain of Foot-and-Mouth Disease Virus Serotype O. Emerg. Infect Dis. 2005, 11, 1887–1893. [Google Scholar] [CrossRef]
- Arzt, J.; Brito, B.; Pauszek, S.J.; Hartwig, E.J.; Smoliga, G.R.; Vu, L.T.; Vu, P.P.; Stenfeldt, C.; Rodriguez, L.L.; Long, N.T.; et al. Genome Sequence of Foot-and-Mouth Disease Virus Serotype O Lineage Ind-2001d Collected in Vietnam in 2015. Genome Announc. 2017, 5, e00223-17. [Google Scholar] [CrossRef]
- Gunasekara, U.; Bertram, M.R.; Dung, D.H.; Hoang, B.H.; Phuong, N.T.; Hung, V.V.; Long, N.V.; Minh, P.Q.; Vu, L.T.; Dong, P.V.; et al. Use of Slaughterhouses as Sentinel Points for Genomic Surveillance of Foot-and-Mouth Disease Virus in Southern Vietnam. Viruses 2021, 13, 2203. [Google Scholar] [CrossRef]
- Pacheco, J.M.; Arzt, J.; Rodriguez, L.L. Early Events in the Pathogenesis of Foot-and-Mouth Disease in Cattle after Controlled Aerosol Exposure. Vet. J. 2010, 183, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hurtle, W.; Rowland, J.M.; Casteran, K.A.; Bucko, S.M.; Grau, F.R.; Valdazo-González, B.; Knowles, N.J.; King, D.P.; Beckham, T.R.; et al. Development of a Universal RT-PCR for Amplifying and Sequencing the Leader and Capsid-Coding Region of Foot-and-Mouth Disease Virus. J Virol. Methods 2013, 189, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Palinski, R.M.; Bertram, M.R.; Vu, L.T.; Pauszek, S.J.; Hartwig, E.J.; Smoliga, G.R.; Stenfeldt, C.; Fish, I.H.; Hoang, B.H.; Phuong, N.T.; et al. First Genome Sequence of Foot-and-Mouth Disease Virus Serotype O Sublineage Ind2001e from Southern Vietnam. Microbiol. Resour. Announc. 2019, 8, e01424-18. [Google Scholar] [CrossRef] [PubMed]
- Bertram, M.R.; Yadav, S.; Stenfeldt, C.; Delgado, A.; Arzt, J. Extinction Dynamics of the Foot-and-Mouth Disease Virus Carrier State Under Natural Conditions. Front. Vet. Sci. 2020, 7, 276. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. Ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef]
- Riebler, A.; Sørbye, S.; Simpson, D.; Rue, H. An Intuitive Bayesian Spatial Model for Disease Mapping That Accounts for Scaling. Stat. Methods Med. Res. 2016, 25, 1145–1165. [Google Scholar] [CrossRef] [PubMed]
- Fuglstad, G.-A.; Simpson, D.; Lindgren, F.; Rue, H. Constructing Priors That Penalize the Complexity of Gaussian Random Fields. J. Am. Stat. Assoc. 2019, 114, 445–452. [Google Scholar] [CrossRef]
- Arzt, J.; Juleff, N.; Zhang, Z.; Rodriguez, L.L. The Pathogenesis of Foot-and-Mouth Disease I: Viral Pathways in Cattle. Transbound. Emerg. Dis. 2011, 58, 291–304. [Google Scholar] [CrossRef]
- Dohoo, I.; Martin, S.; Stryhn, H. Veterinary Epidemiologic Research; VER Inc.: Glendale, CA, USA, 2009. [Google Scholar]
- Simpson, D.P.; Rue, H.; Martins, T.G.; Riebler, A.; Sørbye, S.H. Penalising Model Component Complexity: A Principled, Practical Approach to Constructing Priors. arXiv 2014, arXiv:1403.4630. [Google Scholar] [CrossRef]
- Bivand, R.; Altman, M.; Anselin, L.; Assunção, R.; Berke, O.; Blanchet, F.G.; Carvalho, M.; Christensen, B.; Chun, Y.; Dormann, C.; et al. spdep: Spatial Dependence: Weighting Schemes, Statistics and Models, R package Version 0.3–32. 2006. Available online: https://r-spatial.github.io/spdep/ (accessed on 8 January 2023).
- Wickham, H.; François, R.; Henry, L.; Müller, K. RStudio Dplyr: A Grammar of Data Manipulation. 2022. Available online: https://dplyr.tidyverse.org/ (accessed on 8 January 2023).
- Wickham, H. RStudio Stringr: Simple, Consistent Wrappers for Common String Operations. 2022. Available online: https://rdrr.io/cran/stringr/ (accessed on 8 January 2023).
- Smith, B.J. Boa: An R Package for MCMC Output Convergence Assessment and Posterior Inference. J. Stat. Softw. 2007, 21, 1–37. [Google Scholar] [CrossRef]
- Garnier, S.; Ross, N.; Rudis, B.; Sciaini, M.; Camargo, A.P.; Scherer, C. Viridis: Colorblind-Friendly Color Maps for R. 2021. Available online: https://sjmgarnier.github.io/viridis/ (accessed on 8 January 2023).
- Kassambara, A. Ggpubr: “ggplot2” Based Publication Ready Plots. 2022. Available online: https://rpkgs.datanovia.com/ggpubr/ (accessed on 8 January 2023).
- ickham, H.; Hester, J.; Francois, R.; Bryan, J.; Bearrows, S. Read Rectangular Text Data. 2022. Available online: https://cran.r-project.org/web/packages/readr/readr.pdf (accessed on 8 January 2023).
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Rue, H.; Riebler, A.; Sørbye, S.H.; Illian, J.B.; Simpson, D.P.; Lindgren, F.K. Bayesian Computing with INLA: A Review. Annu. Rev. Stat. Its Appl. 2017, 4, 395–421. [Google Scholar] [CrossRef]
- Baquero, O.S. INLAOutputs. 2022. Available online: https://github.com/oswaldosantos/INLAOutputs (accessed on 8 January 2023).
- Muwonge, A.; Bessell, P.R.; Porphyre, T.; Motta, P.; Rydevik, G.; Devailly, G.; Egbe, N.F.; Kelly, R.F.; Handel, I.G.; Mazeri, S.; et al. Inferring Livestock Movement Networks from Archived Data to Support Infectious Disease Control in Developing Countries. bioRxiv 2021. [Google Scholar] [CrossRef]
- Makau, D.; Alkhamis, M.; Paploski, I.; Corzo, C.; Lycett, S.; VanderWaal, K. Integrating Animal Movements with Phylogeography to Model the Spread of PRRS Virus in the U.S. Virus Evol. 2021, 7, veab060. [Google Scholar] [CrossRef] [PubMed]
- Do, H.; Nguyen, H.-T.-M.; Van Ha, P.; Kompas, T.; Van, K.D.; Chu, L. Estimating the Transmission Parameters of Foot-and-Mouth Disease in Vietnam: A Spatial-Dynamic Kernel-Based Model with Outbreak and Host Data. Prev. Vet. Med. 2022, 208, 105773. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Group | Temporal Effect | Lineage | Cutoff | DIC |
|---|---|---|---|---|
| Spatial | Random effect only | NA | NA | 113457.7 |
| Random effect and random walk | NA | NA | 112329.1 | |
| Phylogeographic | Random effect and random walk | Mya 98 | 0.67 | 110658.8 |
| Random effect and random walk | Mya 98 | 0.55 | 110658.8 | |
| Random effect and random walk | Pan Asia | 0.60 | 110607.7 | |
| Random effect and random walk | Pan Asia | 0.47 | 110623.9 | |
| Random effect and random walk | Total sequences | 0.41 | 110622.9 | |
| Random effect and random walk | Total sequences | 0.37 | 110622.9 | |
| Phylogeographic and spatial | Random effect and random walk | Mya 98 | 0.67 | 112450.0 |
| Random effect and random walk | Mya 98 | 0.55 | 112450.0 | |
| Random effect and random walk | PanAsia | 0.60 | 112710.0 | |
| Random effect and random walk | PanAsia | 0.47 | 112464.6 | |
| Random effect and random walk | Total sequences | 0.41 | 112631.6 | |
| Random effect and random walk | Total sequences | 0.37 | 112631.6 | |
| Phylogeographic and spatial (joint) | Random effect and random walk | Pan Asia | NA | 110611.3 |
| Fixed Effect | Coefficient (95% Credible Interval) |
|---|---|
| Intercept | −2.56 (−3.49, −1.65) |
| Presence of slaughterhouse (ref: no slaughterhouse) | −4.73 (−7.09, −2.55) |
| International border (ref: no international border) | 0.82 (0.18, 1.48) |
| Fixed Effect | Coefficient 95% Credible Interval |
|---|---|
| Intercept | −1.35 (−1.53, −1.35) |
| Pig density | −0.58 (−0.63, −0.53) |
| Presence of Slaughterhouse (ref: no slaughterhouse) | −2.70 (−2.88, −2.51) |
| International border (ref: no international border) | 0.38 (0.34, 0.42) |
| Inferred viral movement from China | −0.57 (−0.68, −0.49) |
| Inferred viral movement from Malaysia | 0.79 (0.64, 0.94) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gunasekara, U.; Bertram, M.R.; Van Long, N.; Minh, P.Q.; Chuong, V.D.; Perez, A.; Arzt, J.; VanderWaal, K. Phylogeography as a Proxy for Population Connectivity for Spatial Modeling of Foot-and-Mouth Disease Outbreaks in Vietnam. Viruses 2023, 15, 388. https://doi.org/10.3390/v15020388
Gunasekara U, Bertram MR, Van Long N, Minh PQ, Chuong VD, Perez A, Arzt J, VanderWaal K. Phylogeography as a Proxy for Population Connectivity for Spatial Modeling of Foot-and-Mouth Disease Outbreaks in Vietnam. Viruses. 2023; 15(2):388. https://doi.org/10.3390/v15020388
Chicago/Turabian StyleGunasekara, Umanga, Miranda R. Bertram, Nguyen Van Long, Phan Quang Minh, Vo Dinh Chuong, Andres Perez, Jonathan Arzt, and Kimberly VanderWaal. 2023. "Phylogeography as a Proxy for Population Connectivity for Spatial Modeling of Foot-and-Mouth Disease Outbreaks in Vietnam" Viruses 15, no. 2: 388. https://doi.org/10.3390/v15020388
APA StyleGunasekara, U., Bertram, M. R., Van Long, N., Minh, P. Q., Chuong, V. D., Perez, A., Arzt, J., & VanderWaal, K. (2023). Phylogeography as a Proxy for Population Connectivity for Spatial Modeling of Foot-and-Mouth Disease Outbreaks in Vietnam. Viruses, 15(2), 388. https://doi.org/10.3390/v15020388