
Patterns and Temporal Dynamics of Natural Recombination in Noroviruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Preparation
2.2. Recombination Analysis
2.3. Phylogenetic Analysis
2.4. Calculation of Recombination Half-Lives
3. Results
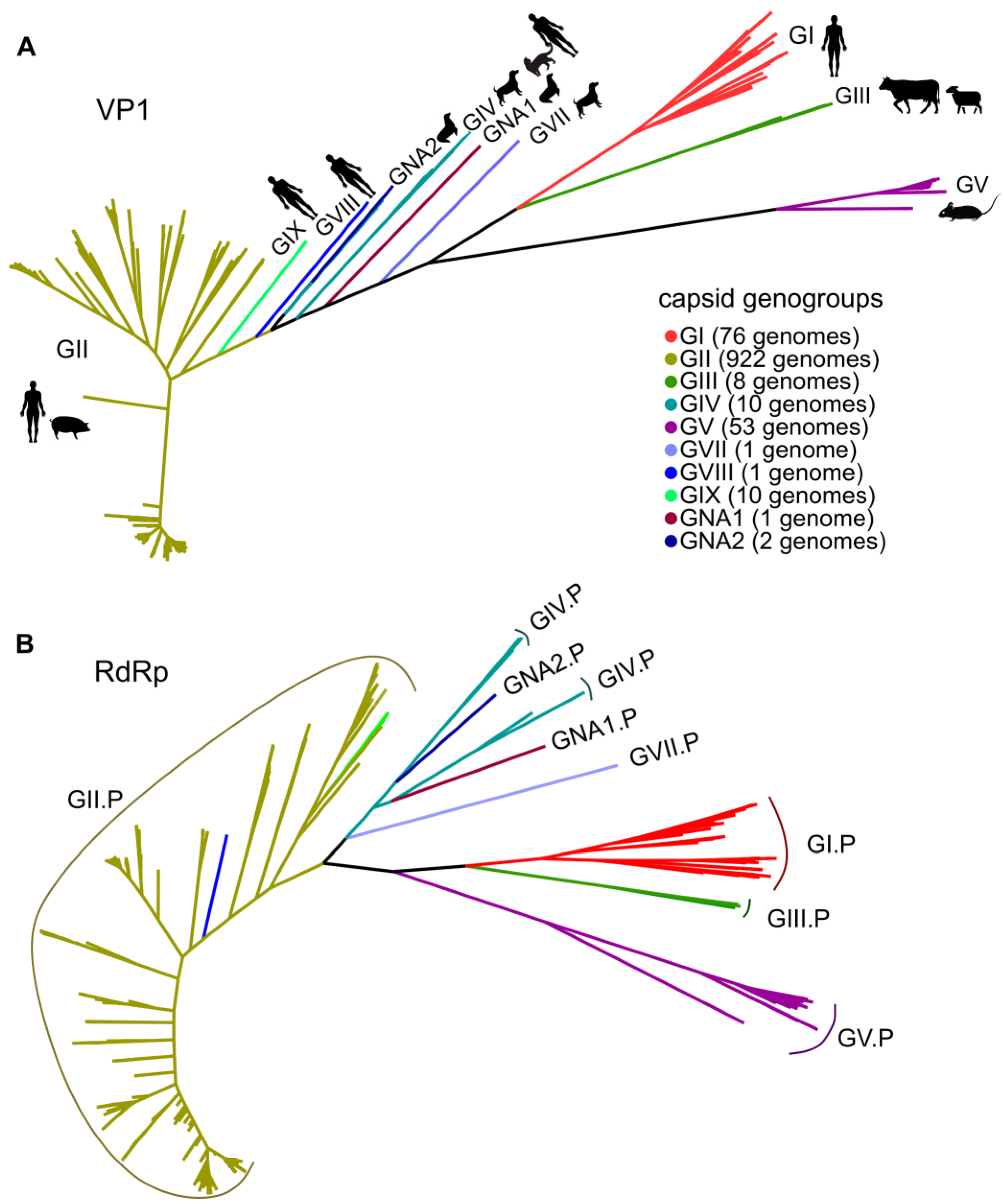
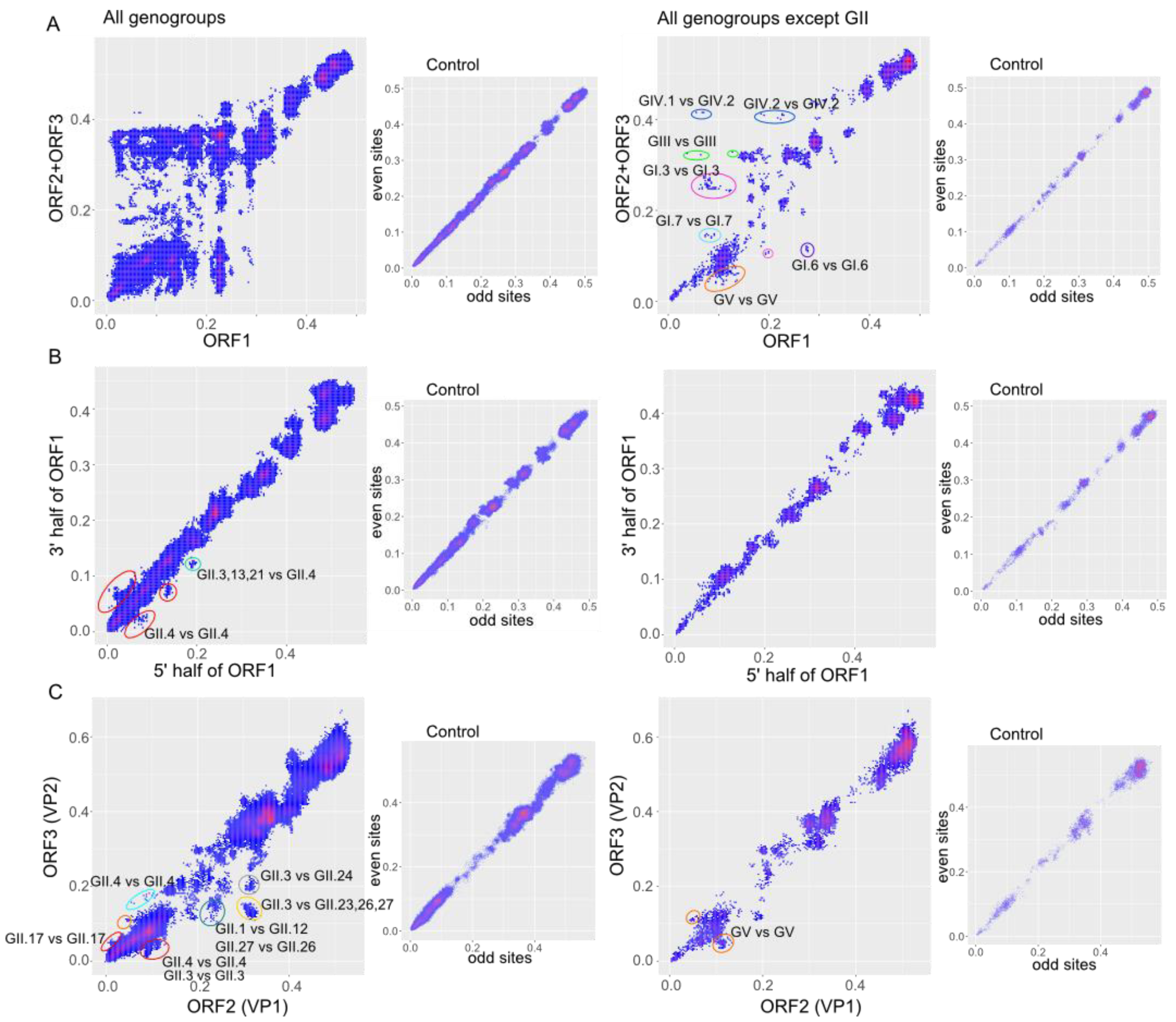
3.1. General Patterns of Recombination in Noroviruses
3.2. Recombination Breakpoints throughout the Genome and among Genogroups
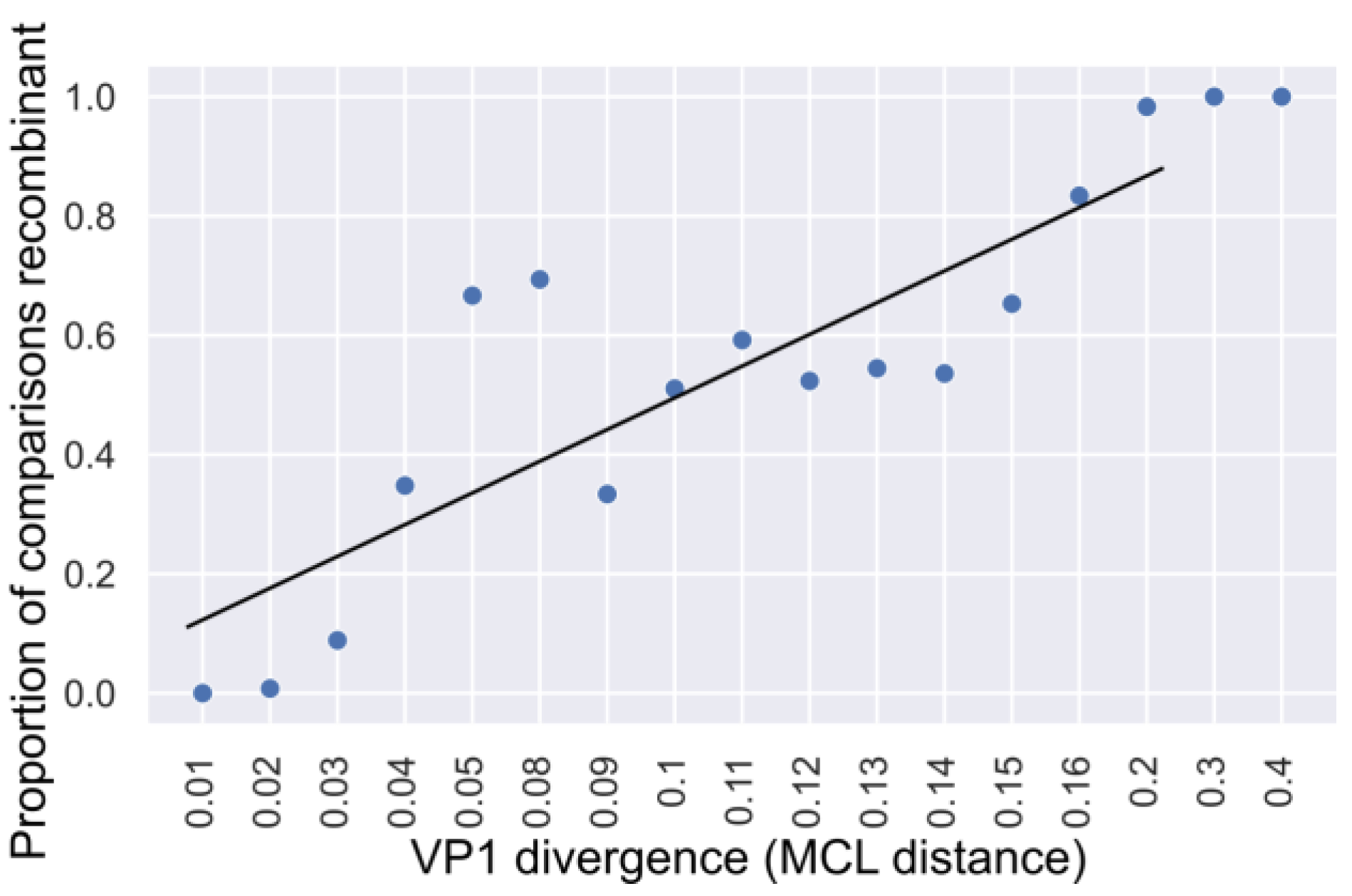
3.3. Genetic Divergence of Noroviruses Involved in Recombination
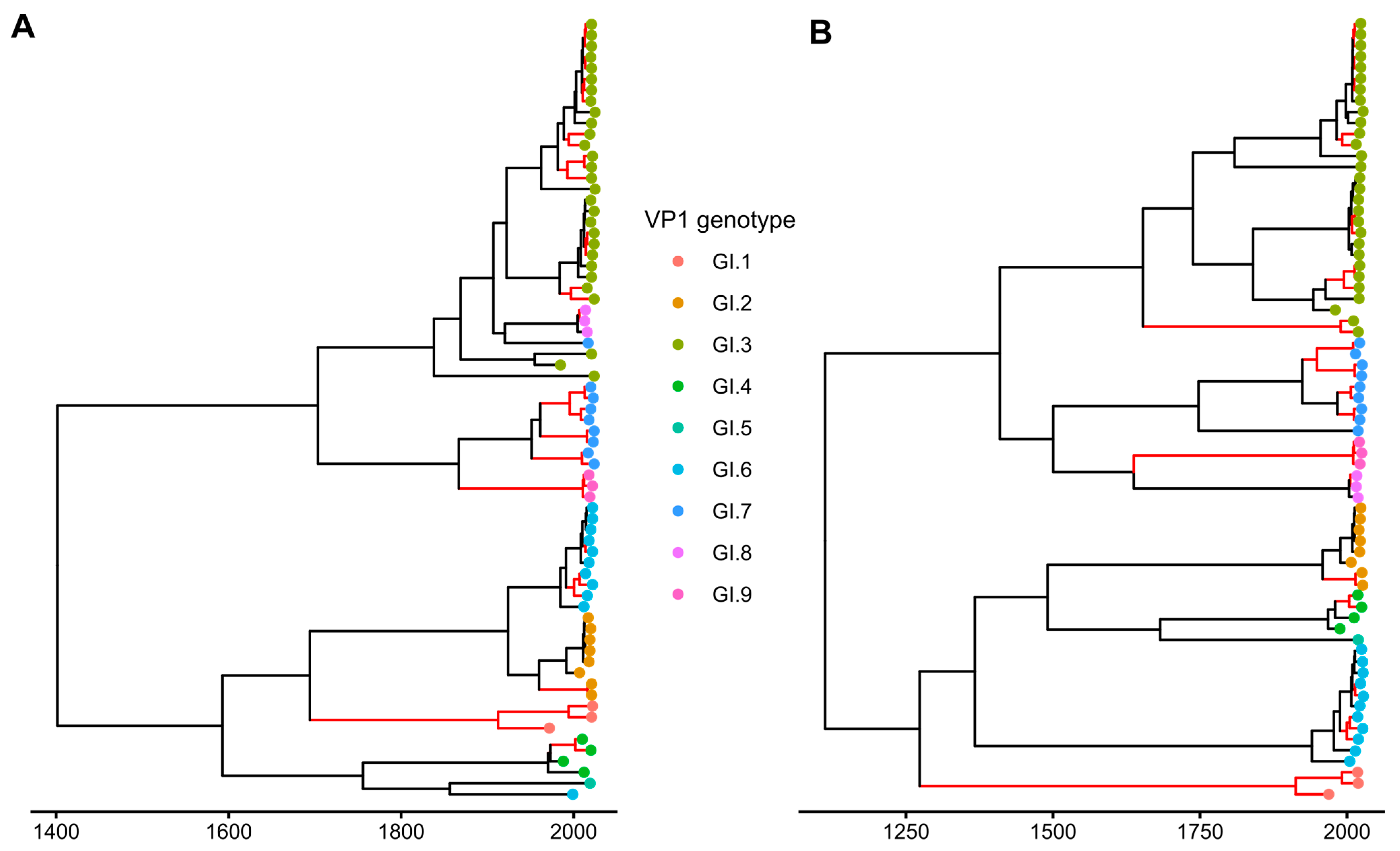
3.4. Temporal Aspects of Norovirus Recombination
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bányai, K.; Estes, M.K.; Martella, V.; Parashar, U.D. Viral gastroenteritis. Lancet 2018, 392, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Green, K. Caliciviridae: The noroviruses. In Fields Virology, 6th ed.; LWW: Philadelphia, PA, USA, 2013; pp. 582–608. [Google Scholar]
- McCormick, C.J.; Salim, O.; Lambden, P.R.; Clarke, I.N. Translation Termination Reinitiation between Open Reading Frame 1 (ORF1) and ORF2 Enables Capsid Expression in a Bovine Norovirus without the Need for Production of Viral Subgenomic RNA. J. Virol. 2008, 82, 8917–8921. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.W.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Kroneman, A.; Vega, E.; Vennema, H.; Vinjé, J.; White, P.A.; Hansman, G.; Green, K.; Martella, V.; Katayama, K.; Koopmans, M. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 2013, 158, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Hardy, M.E.; Kramer, S.F.; Treanor, J.J.; Estes, M.K. Human calicivirus genogroup II capsid sequence diversity revealed by analyses of the prototype Snow Mountain agent. Arch. Virol. 1997, 142, 1469–1479. [Google Scholar] [CrossRef]
- Jiang, X.; Espul, C.; Zhong, W.M.; Cuello, H.; Matson, D.O. Characterization of a novel human calicivirus that may be a naturally occurring recombinant. Arch. Virol. 1999, 144, 2377–2387. [Google Scholar] [CrossRef]
- Begall, L.F.L.; Mauroy, A.; Thiry, E. Norovirus recombinants: Recurrent in the field, recalcitrant in the lab—A scoping review of recombination and recombinant types of noroviruses. J. Gen. Virol. 2018, 99, 970–988. [Google Scholar] [CrossRef]
- Nayak, M.K.; Balasubramanian, G.; Sahoo, G.C.; Bhattacharya, R.; Vinje, J.; Kobayashi, N.; Sarkar, M.C.; Bhattacharya, M.K.; Krishnan, T. Detection of a novel intergenogroup recombinant Norovirus from Kolkata, India. Virology 2008, 377, 117–123. [Google Scholar] [CrossRef]
- Phan, T.G.; Kaneshi, K.; Ueda, Y.; Nakaya, S.; Nishimura, S.; Yamamoto, A.; Sugita, K.; Takanashi, S.; Okitsu, S.; Ushijima, H. Genetic heterogeneity, evolution, and recombination in noroviruses. J. Med. Virol. 2007, 79, 1388–1400. [Google Scholar] [CrossRef]
- Takano, T.; Kusuhara, H.; Kuroishi, A.; Takashina, M.; Doki, T.; Nishinaka, T.; Hohdatsu, T. Molecular characterization and pathogenicity of a genogroup GVI feline norovirus. Vet. Microbiol. 2015, 178, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, B.; Di Profio, F.; Melegari, I.; Sarchese, V.; Cafiero, M.A.; Robetto, S.; Aste, G.; Lanave, G.; Marsilio, F.; Martella, V. A novel feline norovirus in diarrheic cats. Infect. Genet. Evol. 2016, 38, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Rohayem, J.; Münch, J.; Rethwilm, A. Evidence of Recombination in the Norovirus Capsid Gene. J. Virol. 2005, 79, 4977–4990. [Google Scholar] [CrossRef]
- Eden, J.-S.; Tanaka, M.M.; Boni, M.F.; Rawlinson, W.D.; White, P.A. Recombination within the Pandemic Norovirus GII.4 Lineage. J. Virol. 2013, 87, 6270–6282. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.Y.; Zhu, H.; Smith, D.K.; Guan, Y.; Holmes, E.C.; Pybus, O.G. The recombinant origin of emerging human norovirus GII.4/2008: Intra-genotypic exchange of the capsid P2 domain. J. Gen. Virol. 2012, 93, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.; Coughlan, S.; Hall, W.W. Characterisation of a novel recombination event in the norovirus polymerase gene. Virology 2007, 363, 11–14. [Google Scholar] [CrossRef]
- Laconi, A.; Cavicchio, L.; Tassoni, L.; Cunial, G.; Milani, A.; Ustulin, M.; Di Martino, G.; Forzan, M.; Campalto, M.; Monne, I.; et al. Identification of two divergent swine Noroviruses detected at the slaughterhouse in North East Italy. Porc. Health Manag. 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, P.; Walimbe, A.M.; Chitambar, S.D. Complete genome characterization of Genogroup II norovirus strains from India: Evidence of recombination in ORF2/3 overlap. Infect. Genet. Evol. 2010, 10, 1101–1109. [Google Scholar] [CrossRef]
- Ford-Siltz, L.A.; Mullis, L.; Sanad, Y.M.; Tohma, K.; Lepore, C.J.; Azevedo, M.; Parra, G.I. Genomics analyses of GIV and GVI noroviruses reveal the distinct clustering of human and animal viruses. Viruses 2019, 11, 204. [Google Scholar] [CrossRef]
- Thackray, L.B.; Wobus, C.E.; Chachu, K.A.; Liu, B.; Alegre, E.R.; Henderson, K.S.; Kelley, S.T.; Virgin, H.W. Murine Noroviruses Comprising a Single Genogroup Exhibit Biological Diversity despite Limited Sequence Divergence. J. Virol. 2007, 81, 10460–10473. [Google Scholar] [CrossRef]
- Müller, B.; Klemm, U.; Mas Marques, A.; Schreier, E. Genetic diversity and recombination of murine noroviruses in immunocompromised mice. Arch. Virol. 2007, 152, 1709–1719. [Google Scholar] [CrossRef] [PubMed]
- Mathijs, E.; Muylkens, B.; Mauroy, A.; Ziant, D.; Delwiche, T.; Thiry, E. Experimental evidence of recombination in murine noroviruses. J. Gen. Virol. 2010, 91, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cockrell, S.K.; Kolawole, A.O.; Rotem, A.; Serohijos, A.W.R.; Chang, C.B.; Tao, Y.; Mehoke, T.S.; Han, Y.; Lin, J.S.; et al. Isolation and Analysis of Rare Norovirus Recombinants from Coinfected Mice Using Drop-Based Microfluidics. J. Virol. 2015, 89, 7722–7734. [Google Scholar] [CrossRef] [PubMed]
- Martella, V.; Decaro, N.; Lorusso, E.; Radogna, A.; Moschidou, P.; Amorisco, F.; Lucente, M.S.; Desario, C.; Mari, V.; Elia, G.; et al. Genetic Heterogeneity and Recombination in Canine Noroviruses. J. Virol. 2009, 83, 11391–11396. [Google Scholar] [CrossRef]
- McWilliam Leitch, E.C.; Bendig, J.; Cabrerizo, M.; Cardosa, J.; Hyypia, T.; Ivanova, O.E.; Kelly, A.; Kroes, A.C.M.; Lukashev, A.; MacAdam, A.; et al. Transmission Networks and Population Turnover of Echovirus 30. J. Virol. 2009, 83, 2109–2118. [Google Scholar] [CrossRef]
- McWilliam Leitch, E.C.; Cabrerizo, M.; Cardosa, J.; Harvala, H.; Ivanova, O.E.; Kroes, A.C.M.; Lukashev, A.; Muir, P.; Odoom, J.; Roivainen, M.; et al. Evolutionary dynamics and temporal/geographical correlates of recombination in the human enterovirus echovirus types 9, 11, and 30. J. Virol. 2010, 84, 9292–9300. [Google Scholar] [CrossRef]
- McWilliam Leitch, E.C.; Cabrerizo, M.; Cardosa, J.; Harvala, H.; Ivanova, O.E.; Koike, S.; Kroes, A.C.M.; Lukashev, A.; Perera, D.; Roivainen, M.; et al. The association of recombination events in the founding and emergence of subgenogroup evolutionary lineages of human enterovirus 71. J. Virol. 2012, 86, 2676–2685. [Google Scholar] [CrossRef]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kroneman, A.; Vennema, H.; Deforche, K.; Avoort, H.v.d.; Peñaranda, S.; Oberste, M.S.; Vinjé, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Welch, J. Frequency and Dynamics of Recombination within Different Species of Human Enteroviruses Frequency and Dynamics of Recombination within Different Species of Human Enteroviruses. J. Virol. 2006, 80, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, I.B.; Easteal, S. A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Bioinformatics 1996, 12, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Sawyer, S. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 1989, 6, 526–538. [Google Scholar]
- Salminen, M.O.; Carr, J.K.; Burke, D.S.; Mccutchan, F.E. Identification of Breakpoints in Intergenotypic Recombinants of HIV Type 1 by Bootscanning. AIDS Res. Hum. Retroviruses 1995, 11, 1423–1425. [Google Scholar] [CrossRef]
- Smith, J. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in rebombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Weiller, G.F. Phylogenetic profiles: A graphical method for detecting genetic recombinations in homologous sequences. Mol. Biol. Evol. 1998, 15, 326–335. [Google Scholar] [CrossRef]
- Holmes, E.C.; Worobey, M.; Rambaut, A. Phylogenetic evidence for recombination in dengue virus. Mol. Biol. Evol. 1999, 16, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Vakulenko, Y.; Deviatkin, A.; Drexler, J.F.; Lukashev, A. Modular Evolution of Coronavirus Genomes. Viruses 2021, 13, 1270. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Hill, V.; Baele, G. Bayesian Estimation of Past Population Dynamics in BEAST 1.10 Using the Skygrid Coalescent Model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.; Xie, D.; Baele, G.; Suchard, M. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Lukashev, A.N.; Shumilina, E.Y.; Belalov, I.S.; Ivanova, O.E.; Eremeeva, T.P.; Reznik, V.I.; Trotsenko, O.E.; Drexler, J.F.; Drosten, C. Recombination strategies and evolutionary dynamics of the Human enterovirus A global gene pool. J. Gen. Virol. 2014, 95, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yoshizumi, S.; Kogawa, S.; Takahashi, T.; Ueki, Y.; Shinohara, M.; Mizukoshi, F.; Tsukagoshi, H.; Sasaki, Y.; Suzuki, R.; et al. Molecular Evolution of the Capsid Gene in Norovirus Genogroup i. Sci. Rep. 2015, 5, 3806. [Google Scholar] [CrossRef]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.J.; Sosnovtsev, S.V.; Green, K.Y. Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef]
- Kobayashi, M.; Matsushima, Y.; Motoya, T.; Sakon, N.; Shigemoto, N.; Okamoto-Nakagawa, R.; Nishimura, K.; Yamashita, Y.; Kuroda, M.; Saruki, N.; et al. Molecular evolution of the capsid gene in human norovirus genogroup II. Sci. Rep. 2016, 6, 29400. [Google Scholar] [CrossRef]
- Bull, R.A.; Tanaka, M.M.; White, P.A. Norovirus recombination. J. Gen. Virol. 2007, 88, 3347–3359. [Google Scholar] [CrossRef]
- Kendra, J.A.; Tohma, K.; Parra, G.I. Global and regional circulation trends of norovirus genotypes and recombinants, 1995–2019: A comprehensive review of sequences from public databases. Rev. Med. Virol. 2022, 32, e2354. [Google Scholar] [CrossRef]
- Ludwig-Begall, L.F.; Mauroy, A.; Thiry, E. Noroviruses—The State of the Art, Nearly Fifty Years after Their Initial Discovery. Viruses 2021, 13, 1541. [Google Scholar] [CrossRef]
- Bull, R.A.; Hansman, G.S.; Clancy, L.E.; Tanaka, M.M.; Rawlinson, W.D.; White, P.A. Norovirus recombination in ORF1/ORF2 overlap. Emerg. Infect. Dis. 2005, 11, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Kulka, M.; Coughlan, S.; Green, K.Y.; Parra, G.I. Genomic analyses of human sapoviruses detected over a 40-year period reveal disparate patterns of evolution among genotypes and genome regions. Viruses 2020, 12, 516. [Google Scholar] [CrossRef] [PubMed]
- Gmyl, A.P.; Belousov, E.V.; Maslova, S.V.; Khitrina, E.V.; Chetverin, A.B.; Agol, V.I. Nonreplicative RNA Recombination in Poliovirus. J. Virol. 1999, 73, 8958–8965. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.; Evans, D.J. Mechanisms and consequences of positive-strand RNA virus recombination. J. Gen. Virol. 2018, 99, 1345–1356. [Google Scholar] [CrossRef]
- Runckel, C.; Westesson, O.; Andino, R.; DeRisi, J.L. Identification and Manipulation of the Molecular Determinants Influencing Poliovirus Recombination. PLoS Pathog. 2013, 9, e1003164. [Google Scholar] [CrossRef]
- Romanova, L.I.; Blinov, V.M.; Tolskaya, E.A.; Viktorova, E.G.; Kolesnikova, M.S.; Guseva, E.A.; Agol, V.I. The primary structure of crossover regions of intertypic poliovirus recombinants: A model of recombination between RNA genomes. Virology 1986, 155, 202–213. [Google Scholar] [CrossRef]
- Blundell, R.J. An Investigation into Genome-Scale Ordered RNA Structure (GORS) in Murine Norovirus and Other Positive-Stranded RNA Viruses. Ph.D. Thesis, Edinburgh Medical School, Scotland, UK, 2010. [Google Scholar]
- Lukashev, A. Recombination among picornaviruses. Rev. Med. Virol. 2010, 20, 327–337. [Google Scholar] [CrossRef]
- Simmonds, P. Recombination and selection in the evolution of picornaviruses and other Mammalian positive-stranded RNA viruses. J. Virol. 2006, 80, 11124–11140. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vakulenko, Y.A.; Orlov, A.V.; Lukashev, A.N. Patterns and Temporal Dynamics of Natural Recombination in Noroviruses. Viruses 2023, 15, 372. https://doi.org/10.3390/v15020372
Vakulenko YA, Orlov AV, Lukashev AN. Patterns and Temporal Dynamics of Natural Recombination in Noroviruses. Viruses. 2023; 15(2):372. https://doi.org/10.3390/v15020372
Chicago/Turabian StyleVakulenko, Yulia A., Artem V. Orlov, and Alexander N. Lukashev. 2023. "Patterns and Temporal Dynamics of Natural Recombination in Noroviruses" Viruses 15, no. 2: 372. https://doi.org/10.3390/v15020372
APA StyleVakulenko, Y. A., Orlov, A. V., & Lukashev, A. N. (2023). Patterns and Temporal Dynamics of Natural Recombination in Noroviruses. Viruses, 15(2), 372. https://doi.org/10.3390/v15020372