
Differential Cellular Sensing of Fusion from within and Fusion from without during Virus Infection
 and
and 
Abstract
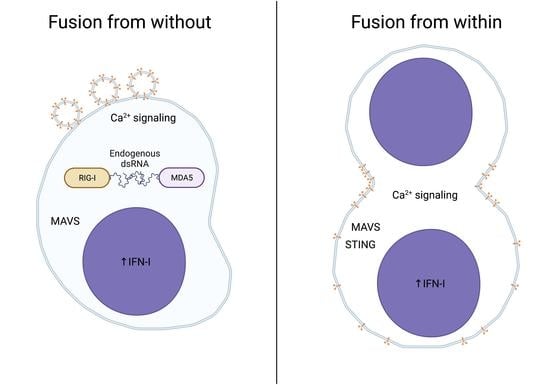
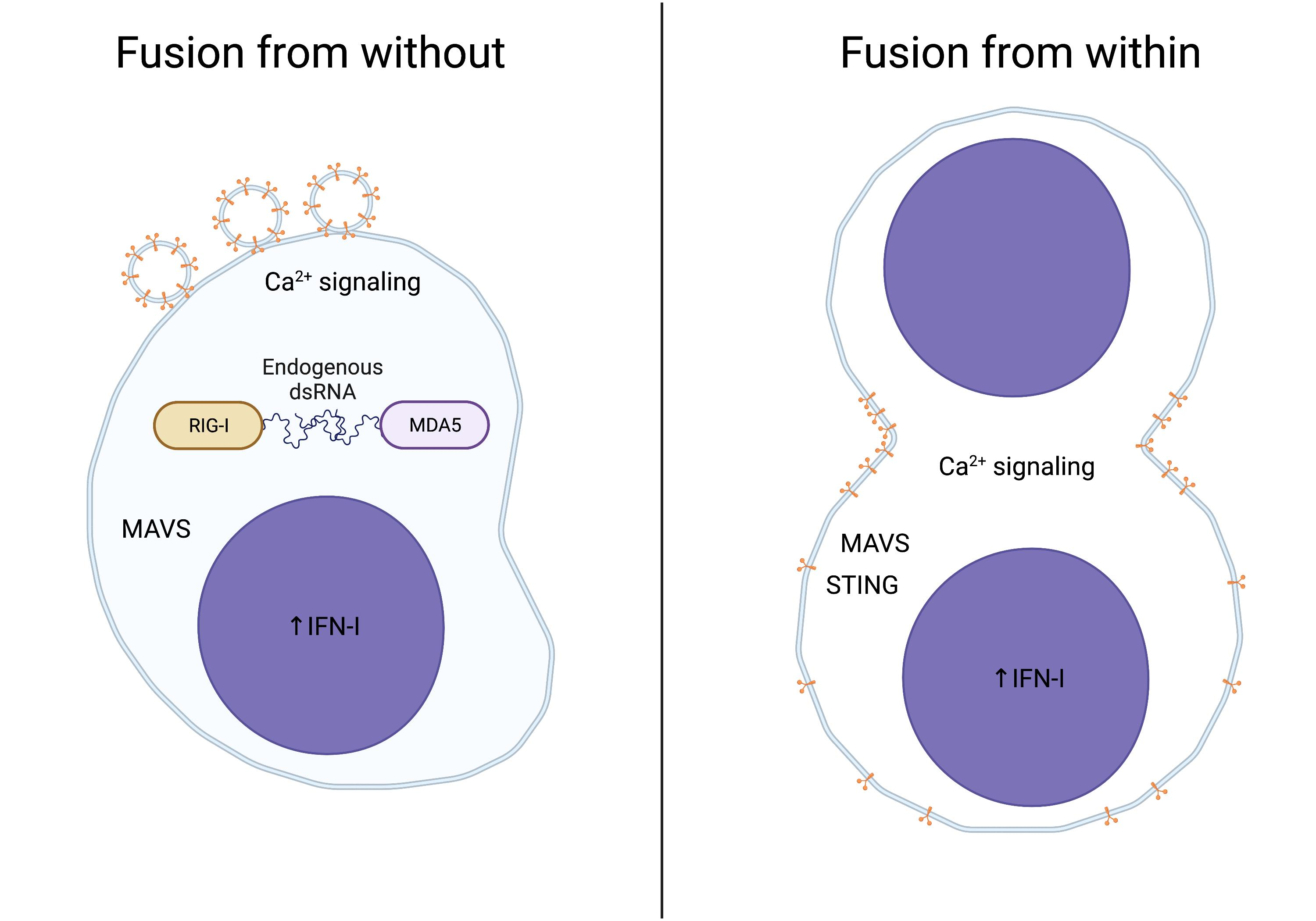{kind=link}
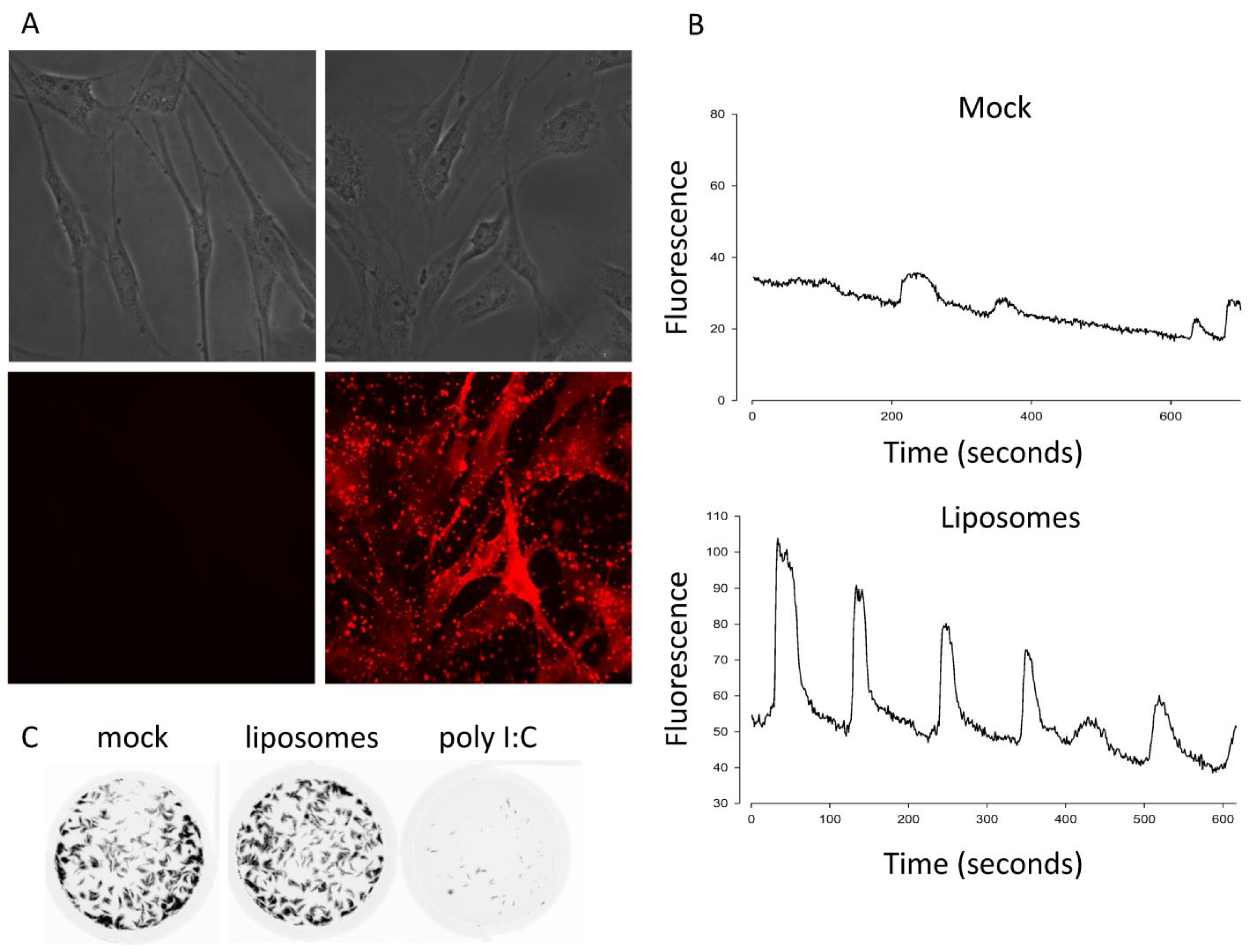{kind=link}
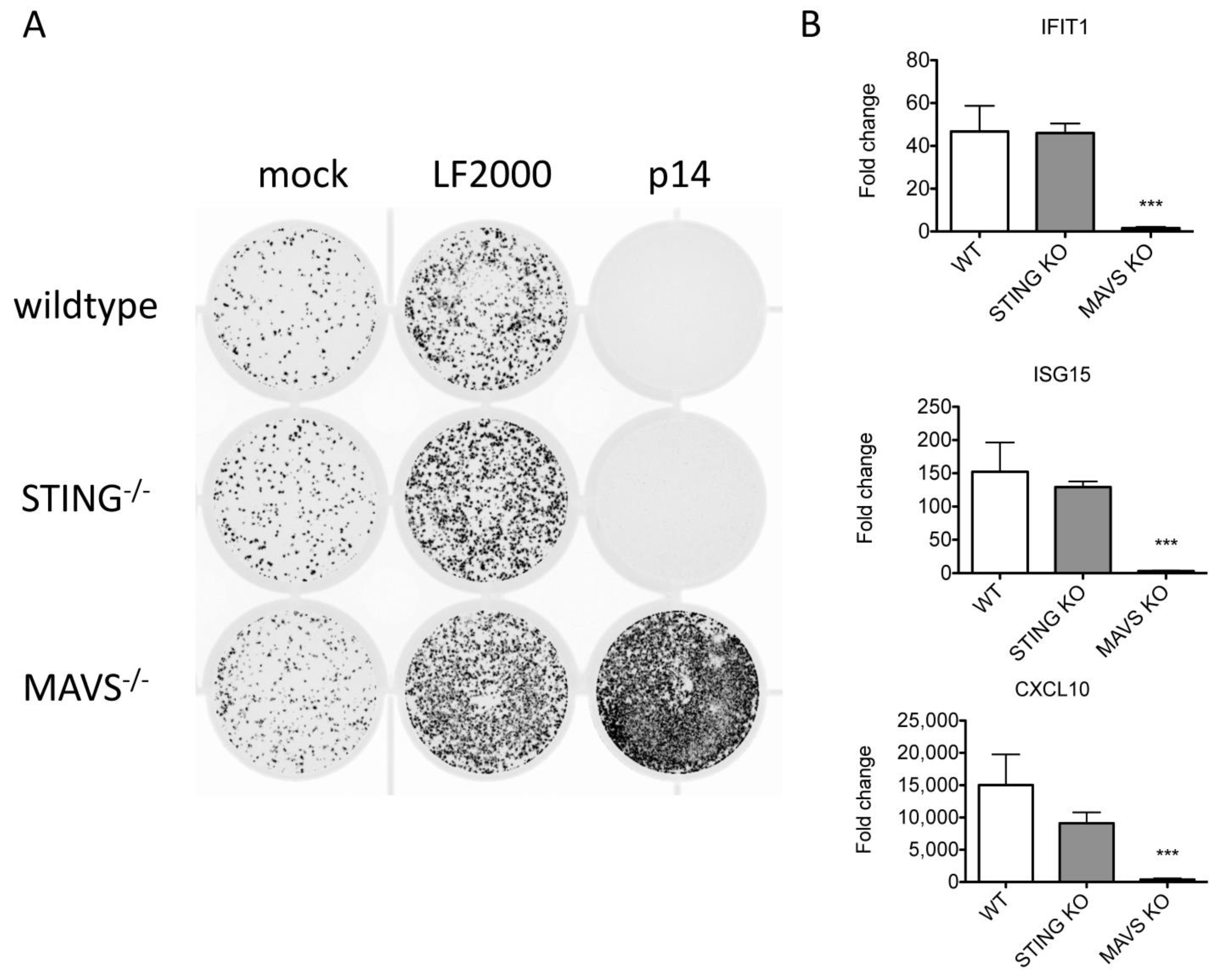{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Fusogenic Liposomes and p14 Lipoplexes
2.3. Generation of Cell Lines
2.4. Quantitative RT-PCR
2.5. Live Cell Fluorescence Microscopy
2.6. Fixed Cell Imaging
2.7. Plaque Reduction Assay
2.8. Western Blotting
2.9. Dot Blots
3. Results
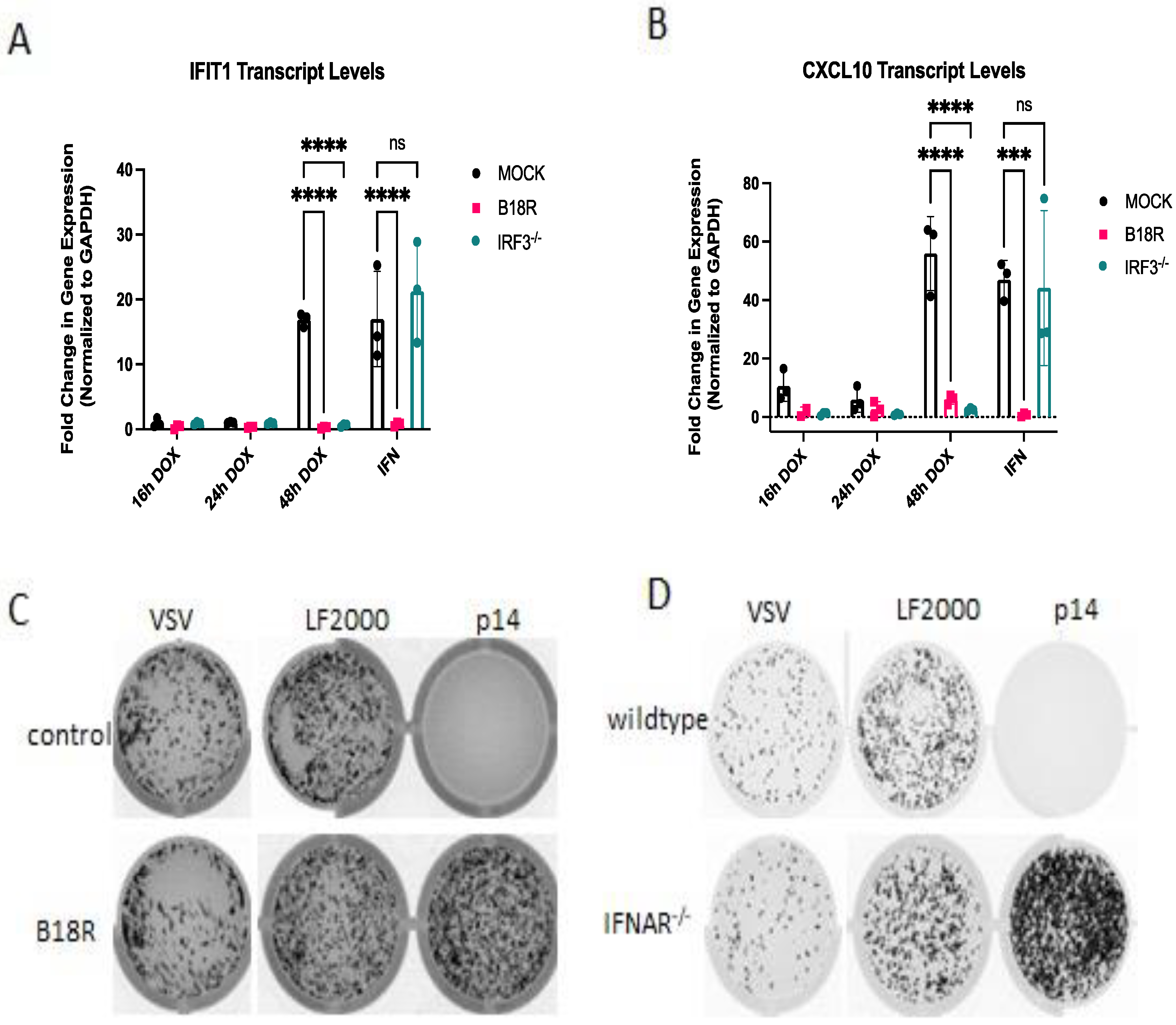
3.1. Calcium Is Necessary but Not Sufficient for Antivial State Induction
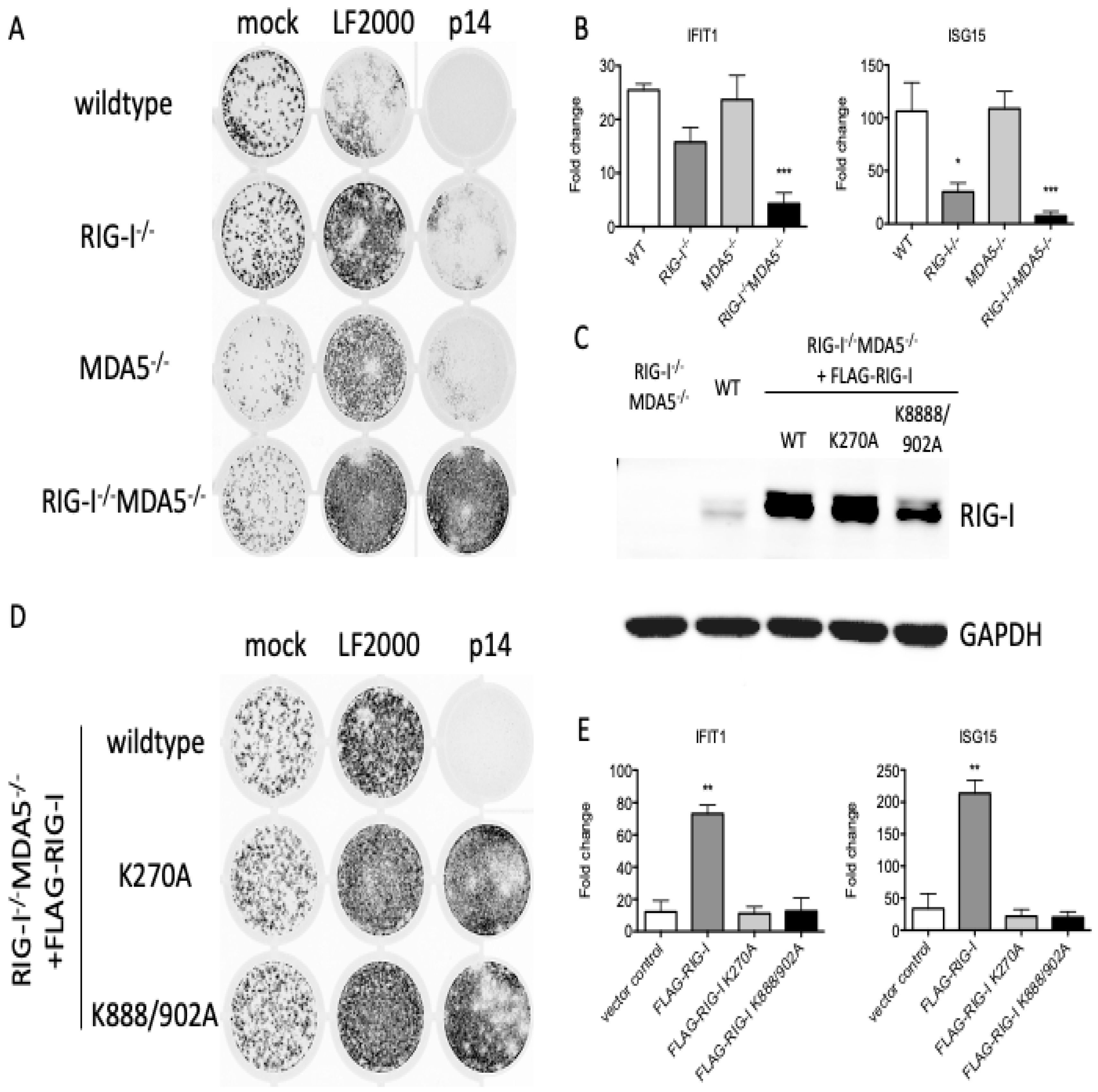
3.2. RNA Sensors Are Required for the Response to Exogenous p14
3.3. The Response to p14 Requires the RNA Binding Sites of RIG-I
3.4. The Response to p14 Is Not Due to Contaminating RNA
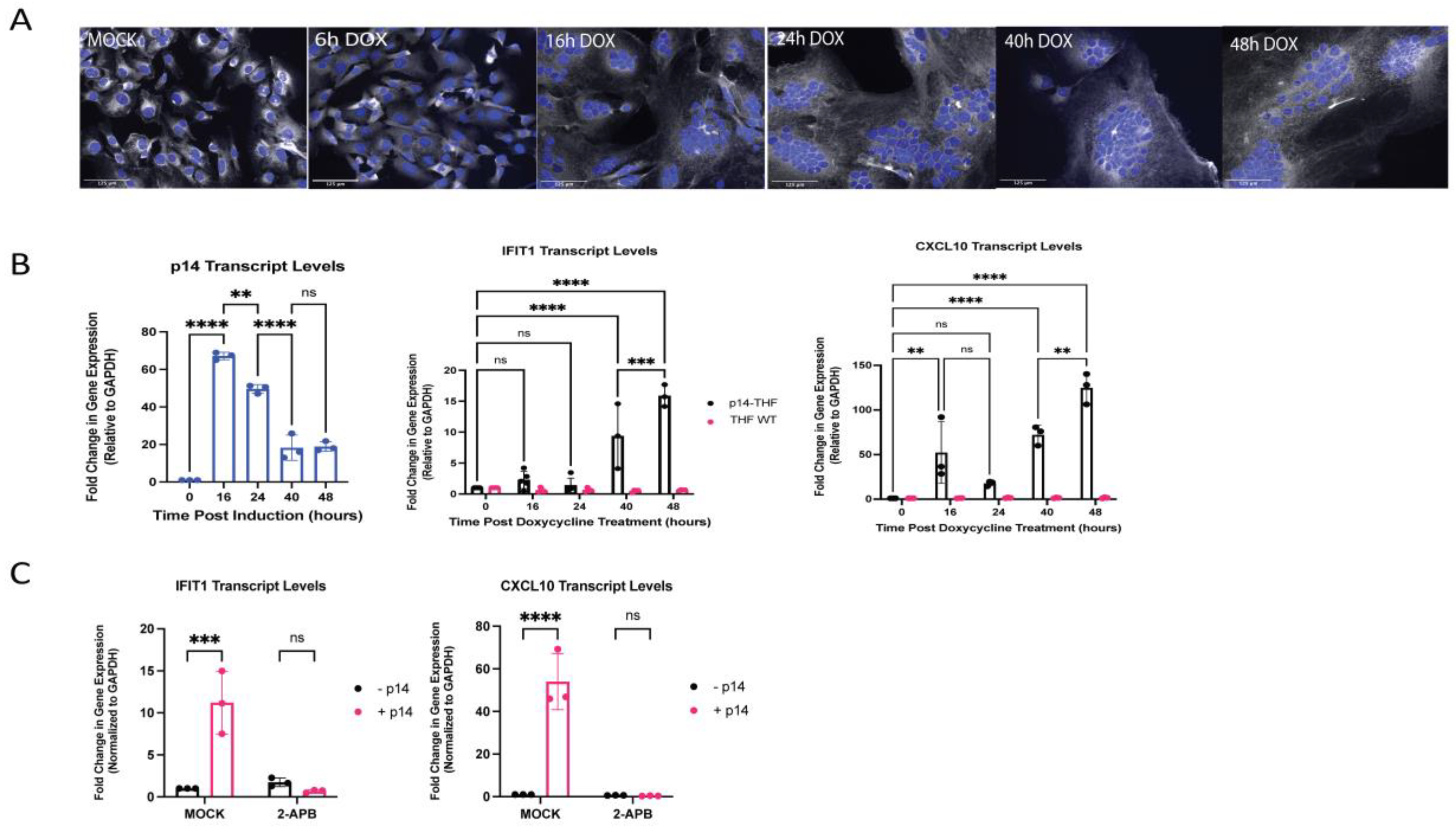
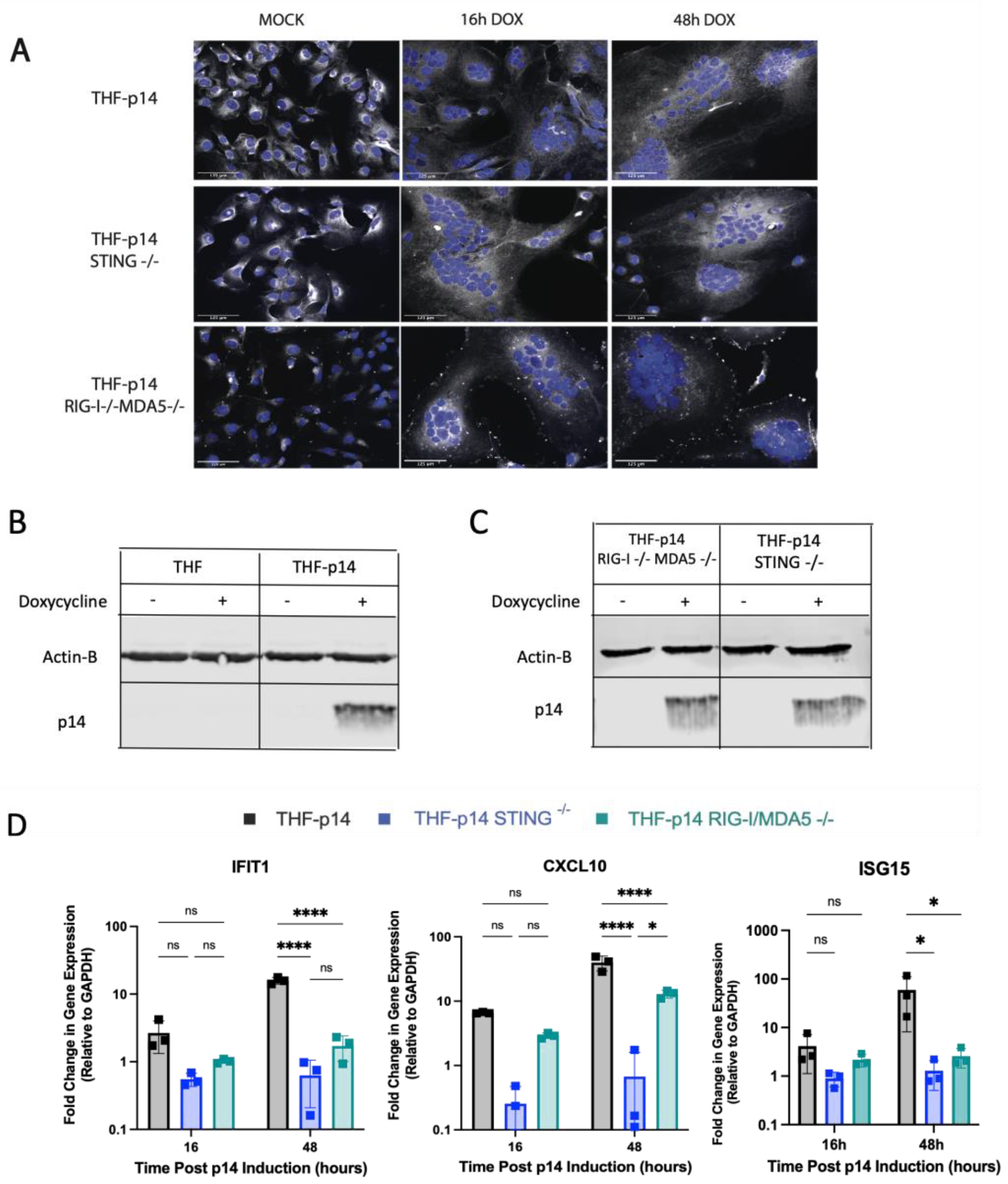
3.5. Generation of Endogenous p14-Expressing Cell Lines to Model Cell–Cell Fusion
3.6. The ISG Response to Endogenous p14 Requires Both STING and RIG-I
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muller, U.; Steinhoff, U.; Reis, L.F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 2016, 16, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef]
- Juckem, L.K.; Boehme, K.W.; Feire, A.L.; Compton, T. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J. Immunol. 2008, 180, 4965–4977. [Google Scholar] [CrossRef]
- Colaco, H.G.; Moita, L.F. Initiation of innate immune responses by surveillance of homeostasis perturbations. FEBS J. 2016, 283, 2448–2457. [Google Scholar] [CrossRef]
- Hare, D.; Mossman, K.L. Novel paradigms of innate immune sensing of viral infections. Cytokine 2013, 63, 219–224. [Google Scholar] [CrossRef]
- Collins, S.E.; Mossman, K.L. Danger, diversity and priming in innate antiviral immunity. Cytokine Growth Factor Rev. 2014, 25, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Roers, A.; Hiller, B.; Hornung, V. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 2016, 44, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Noyce, R.S.; Taylor, K.; Ciechonska, M.; Collins, S.E.; Duncan, R.; Mossman, K.L. Membrane perturbation elicits an IRF3-dependent, interferon-independent antiviral response. J. Virol. 2011, 85, 10926–10931. [Google Scholar] [CrossRef]
- Hare, D.N.; Collins, S.E.; Mukherjee, S.; Loo, Y.M.; Gale, M., Jr.; Janssen, L.J.; Mossman, K.L. Membrane Perturbation-Associated Ca2+ Signaling and Incoming Genome Sensing Are Required for the Host Response to Low-Level Enveloped Virus Particle Entry. J. Virol. 2015, 90, 3018–3027. [Google Scholar] [CrossRef] [PubMed]
- Holm, C.K.; Rahbek, S.H.; Gad, H.H.; Bak, R.O.; Jakobsen, M.R.; Jiang, Z.; Hansen, A.L.; Jensen, S.K.; Sun, C.; Thomsen, M.K.; et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat. Commun. 2016, 7, 10680. [Google Scholar] [CrossRef] [PubMed]
- Holm, C.K.; Jensen, S.B.; Jakobsen, M.R.; Cheshenko, N.; Horan, K.A.; Moeller, H.B.; Gonzalez-Dosal, R.; Rasmussen, S.B.; Christensen, M.H.; Yarovinsky, T.O.; et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 2012, 13, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Hersch, N.; Dieluweit, S.; Biehl, R.; Merkel, R.; Hoffmann, B. Novel fusogenic liposomes for fluorescent cell labeling and membrane modification. Bioconjugate Chem. 2010, 21, 537–543. [Google Scholar] [CrossRef]
- Li, Z.; Michael, I.P.; Zhou, D.; Nagy, A.; Rini, J.M. Simple piggyBac transposon-based mammalian cell expression system for inducible protein production. Proc. Natl. Acad. Sci. USA 2013, 110, 5004–5009. [Google Scholar] [CrossRef]
- Bresnahan, W.A.; Hultman, G.E.; Shenk, T. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J. Virol. 2000, 74, 10816–10818. [Google Scholar] [CrossRef]
- Sali, T.M.; Pryke, K.M.; Abraham, J.; Liu, A.; Archer, I.; Broeckel, R.; Staverosky, J.A.; Smith, J.L.; Al-Shammari, A.; Amsler, L.; et al. Characterization of a Novel Human-Specific STING Agonist that Elicits Antiviral Activity Against Emerging Alphaviruses. PLoS Pathog. 2015, 11, e1005324. [Google Scholar] [CrossRef]
- DeWitte-Orr, S.J.; Mehta, D.R.; Collins, S.E.; Suthar, M.S.; Gale, M., Jr.; Mossman, K.L. Long double-stranded RNA induces an antiviral response independent of IFN regulatory factor 3, IFN-beta promoter stimulator 1, and IFN. J. Immunol. 2009, 183, 6545–6553. [Google Scholar] [CrossRef] [PubMed]
- Haan, C.; Behrmann, I. A cost effective non-commercial ECL-solution for Western blot detections yielding strong signals and low background. J. Immunol. Methods 2007, 318, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Stojdl, D.F.; Taylor, R.A.; Miller, L.; Frenkel, I.; Atkins, H.; Bell, J.C. Vesicular stomatitis virus: A potential therapeutic virus for the treatment of hematologic malignancy. Hum. Gene Ther. 2004, 15, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Gack, M.U. Mechanisms of RIG-I-like receptor activation and manipulation by viral pathogens. J. Virol. 2014, 88, 5213–5216. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Takahasi, K.; Yoneyama, M.; Nishihori, T.; Hirai, R.; Kumeta, H.; Narita, R.; Gale, M., Jr.; Inagaki, F.; Fujita, T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 2008, 29, 428–440. [Google Scholar] [CrossRef]
- Collins, S.E.; Noyce, R.S.; Mossman, K.L. Innate cellular response to virus particle entry requires IRF3 but not virus replication. J. Virol. 2004, 78, 1706–1717. [Google Scholar] [CrossRef]
- Shu, C.; Yi, G.; Watts, T.; Kao, C.C.; Li, P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat. Struct. Mol. Biol. 2012, 19, 722–724. [Google Scholar] [CrossRef]
- Srikanth, S.; Woo, J.S.; Wu, B.; El-Sherbiny, Y.M.; Leung, J.; Chupradit, K.; Rice, L.; Seo, G.J.; Calmettes, G.; Ramakrishna, C.; et al. The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 2019, 20, 152–162. [Google Scholar] [CrossRef]
- Wu, J.; Yan, N. STIM1 moonlights as an anchor for STING. Nat. Immunol. 2019, 20, 112–114. [Google Scholar] [CrossRef] [PubMed]
- Hyser, J.M.; Utama, B.; Crawford, S.E.; Broughman, J.R.; Estes, M.K. Activation of the endoplasmic reticulum calcium sensor STIM1 and store-operated calcium entry by rotavirus requires NSP4 viroporin activity. J. Virol. 2013, 87, 13579–13588. [Google Scholar] [CrossRef] [PubMed]
- Mathavarajah, S.; Salsman, J.; Dellaire, G. An emerging role for calcium signalling in innate and autoimmunity via the cGAS-STING axis. Cytokine Growth Factor Rev. 2019, 50, 43–51. [Google Scholar] [CrossRef]
- Hay, J.C. Calcium: A fundamental regulator of intracellular membrane fusion? EMBO Rep. 2007, 8, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Ciechonska, M.; Key, T.; Duncan, R. Efficient reovirus- and measles virus-mediated pore expansion during syncytium formation is dependent on annexin A1 and intracellular calcium. J. Virol. 2014, 88, 6137–6147. [Google Scholar] [CrossRef]
- Ku, J.W.K.; Chen, Y.; Lim, B.J.W.; Gasser, S.; Crasta, K.C.; Gan, Y.H. Bacterial-induced cell fusion is a danger signal triggering cGAS-STING pathway via micronuclei formation. Proc. Natl. Acad. Sci. USA 2020, 117, 15923–15934. [Google Scholar] [CrossRef]
- Corcoran, J.A.; Duncan, R. Reptilian reovirus utilizes a small type III protein with an external myristylated amino terminus to mediate cell-cell fusion. J. Virol. 2004, 78, 4342–4351. [Google Scholar] [CrossRef]
- Chiang, J.J.; Sparrer, K.M.J.; van Gent, M.; Lassig, C.; Huang, T.; Osterrieder, N.; Hopfner, K.P.; Gack, M.U. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 2018, 19, 53–62. [Google Scholar] [CrossRef]
- Kim, S.Y.; Park, C.; Jang, H.J.; Kim, B.O.; Bae, H.W.; Chung, I.Y.; Kim, E.S.; Cho, Y.H. Antibacterial strategies inspired by the oxidative stress and response networks. J. Microbiol. 2019, 57, 203–212. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- Ranoa, D.R.; Parekh, A.D.; Pitroda, S.P.; Huang, X.; Darga, T.; Wong, A.C.; Huang, L.; Andrade, J.; Staley, J.P.; Satoh, T.; et al. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 2016, 7, 26496–26515. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, N.; Mertens, C.; Zillinger, T.; Wenzel, J.; Bald, T.; Zahn, S.; Tuting, T.; Hartmann, G.; Barchet, W. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 2013, 39, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Carletti, T.; Zakaria, M.K.; Faoro, V.; Reale, L.; Kazungu, Y.; Licastro, D.; Marcello, A. Viral priming of cell intrinsic innate antiviral signaling by the unfolded protein response. Nat. Commun. 2019, 10, 3889. [Google Scholar] [CrossRef] [PubMed]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Eckard, S.C.; Rice, G.I.; Fabre, A.; Badens, C.; Gray, E.E.; Hartley, J.L.; Crow, Y.J.; Stetson, D.B. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat. Immunol. 2014, 15, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Hemmerling, I.; Schmid-Burgk, J.L.; Behrendt, R.; Roers, A.; Hornung, V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J. Immunol. 2014, 192, 5993–5997. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhang, X.; Lei, X.; Xiao, X.; Jiao, T.; Ma, R.; Dong, X.; Jiang, Q.; Wang, W.; Shi, Y.; et al. Sensing of cytoplasmic chromatin by cGAS activates innate immune response in SARS-CoV-2 infection. Signal Transduct. Target. Ther. 2021, 6, 382. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hare, D.N.; Murdza, T.; Collins, S.; Schulz, K.; Mukherjee, S.; de Antueno, R.; Janssen, L.; Duncan, R.; Mossman, K.L. Differential Cellular Sensing of Fusion from within and Fusion from without during Virus Infection. Viruses 2023, 15, 301. https://doi.org/10.3390/v15020301
Hare DN, Murdza T, Collins S, Schulz K, Mukherjee S, de Antueno R, Janssen L, Duncan R, Mossman KL. Differential Cellular Sensing of Fusion from within and Fusion from without during Virus Infection. Viruses. 2023; 15(2):301. https://doi.org/10.3390/v15020301
Chicago/Turabian StyleHare, David N., Tetyana Murdza, Susan Collins, Katharina Schulz, Subhendu Mukherjee, Roberto de Antueno, Luke Janssen, Roy Duncan, and Karen L. Mossman. 2023. "Differential Cellular Sensing of Fusion from within and Fusion from without during Virus Infection" Viruses 15, no. 2: 301. https://doi.org/10.3390/v15020301
APA StyleHare, D. N., Murdza, T., Collins, S., Schulz, K., Mukherjee, S., de Antueno, R., Janssen, L., Duncan, R., & Mossman, K. L. (2023). Differential Cellular Sensing of Fusion from within and Fusion from without during Virus Infection. Viruses, 15(2), 301. https://doi.org/10.3390/v15020301