1. Introduction
The Western honey bee,
Apis mellifera (Linnaeus, 1758), is an important pollinator in agriculture, but also in natural ecosystems worldwide [
1,
2]. Due to its pollination of crops and associated honey production, this honey bee species has been spread around the world and has become irreplaceable in some agricultural fields [
3]. The decline in honey bee health observed in recent decades and the recurring winter losses of honey bee colonies in the northern hemisphere are of concern to the wider public [
4,
5,
6]. A combination of several different factors is likely to be the reason for these health problems that have practically wiped out the wild populations of honey bees in northern Europe and North America [
7]. In addition to abiotic factors such as global warming [
8], intensification of agriculture, industrial-scale management practices [
9], and the agricultural use of pesticides [
10], multiple biotic factors play a major role for the well-being of honey bees. Recent studies have shown that parasites and viral pathogens are of outstanding importance for the health status and survival of honey bee colonies [
11]. Following the emergence of the ectoparasite mite
Varroa destructor (Anderson and Trueman, 2000) [
12], the deformed wing virus (DWV) evolved from a minor pathogen into a serious pest in beekeeping [
13]. Varroa mites feed on the fat body of adult honey bees and their capped brood, weakening the host animals and acting as potent vectors for DWV [
14,
15]. The mite bites penetrate the natural barriers of the bee body and inject the virus directly into the hemolymph. DWV infections are usually asymptomatic in adult bees but cause severe disease in the developing brood, resulting in malformation, dwarfism, discoloration, and ultimately death [
16,
17]. However, virus infections have different consequences for the bee colony depending on the number of infected individuals and the severity of disease. So-called covert infections, in which the virus is already detectable in single individuals via molecular diagnostics but no clinical signs are yet observed at a colony level, occur regularly throughout the year [
18]. If the mite infestation rate increases during the summer, so-called overt infections may result. Overt infections are characterized by the appearance of crippled young bees and eventually by the demise of the whole colony [
19]. The outcome of DWV infections depends on different factors, such as the virulence of the respective viral strains [
20], the genetic characteristics of the honey bees and their immunological status [
21], and ecological factors that the colony encounters. It must be emphasized that the burden of varroa mites, which act as a DWV vector, is certainly the most important factor in the development of overt disease and DWV-related colony collapses [
22].
DWV is a member of the genus
Iflavirus and the family
Iflaviridae within the order
Picornavirales [
23]. Four master variants of the virus have been described: DWV-A, -B, -C, and -D [
24]. Like other members of
Picornavirales, DWV forms non-enveloped icosahedral virions, in which the RNA is encapsidated by the structural proteins VP1, VP2, and VP3 [
25]. The non-segmented single-stranded positive-sensed RNA genome has a length of about 10 kb. It harbors a single open reading frame (ORF) encoding one large polyprotein of 2893 amino acids, which is co- and post-translationally processed into the mature proteins [
26]. The initiation of the cap-independent translation is mediated by an IRES element located within the large 5’-UTR of 1167 bps [
27]. While the structural proteins are located at the N-terminal half, the non-structural proteins are located at the C-terminus of the polyprotein. A rather short 3´-UTR with a total length of 308 bps is followed by an mRNA-like poly-A tail. Although DWV is one of the best-studied insect viruses, many aspects of the viral lifecycle are still unknown, such as cellular receptors, host factors involved in RNA replication, and last but not least, the exact processing of the viral polyprotein [
28].
The processing of the polyprotein is mainly mediated by one major protease in the picornavirus-like iflaviruses. This cysteine protease has been designated a 3C in the viruses within the family
Picornaviridae [
29,
30]. It displays a highly conserved catalytic triad consisting of a cysteine, histidine and glutamate, or aspartate residue. The activity of the 3C protease is critical for the processing of the viral polyprotein and is therefore essential for viral replication and genome encapsidation [
31]. In addition to the essential cleavage of viral polypeptides, the different 3C proteases also cleave multiple cellular target proteins to manipulate the host cell. Known cellular targets of 3Cs include factors involved in transcription, translation, and nucleocytoplasmic trafficking [
32]. The 3C thus modulates metabolism and, most importantly, the cellular mRNA transcription and cap-mediated translation to prioritize cap-independent translation of viral genomes via IRES elements [
33,
34]. Unprocessed precursor molecules consisting of the 3C protease and the RNA-dependent RNA polymerase (RdRp, 3D) were shown to be required for the initiation of RNA synthesis and are also proteolytically active [
35,
36]. Based on amino acid sequence alignments and striking secondary structure similarities, comparable 3C-like (3CL) cysteine proteases have been found in all
Picornavirales, including the iflaviruses [
37].
Several studies have already provided insights into the properties of 3CL proteases of iflaviruses. As early as 2012, an active 3CL protease of the Ectropis obliqua virus (EoV) was produced recombinantly, which allowed for the identification of the N- and C-termini of the mature enzyme using autoproteolysis and Edman degradation [
37]. The cleavage site nomenclature designates the carboxyterminus generated by the cleavage of a peptide bond as P1, while the newly generated N-terminus is designated P1′ [
38]. The identified cleavage sites between P1 Q and P1’ S as well as between P1 E and P1’ A had the typical features of the substrates of many known 3C and 3CL proteases, which usually utilize P1 Q/E and P1’ G/S/A cleavage sites [
39]. Ye and colleagues also identified the residues of the catalytic triad within the active center of the EoV protease via an amino acid alignment. They confirmed their prediction via mutational analyses exchanging the amino acids H
2261, D
2299, and C
2383 against alanine residues. While the mutations H
2261A and C
2383A completely eradicated the activity of the recombinant enzyme, the mutation D
2299A resulted in low levels of residual activity. Different cis- and trans-cleavage assays using purified recombinant EoV 3CL protease uncovered basic biochemical properties of the 3CL protease of
Iflaviridae in vitro. Tests of known serine or cysteine protease inhibitors showed that the 3CL protease of EoV is sensitive to p-chloromercuribenzenesulfonate (PCMBS), methyl methanethiosulfonate (MMTS), and N-ethylmaleimide (NEM), as expected for a cysteine protease. Taken together, the authors were able to provide a complete characterization of the EoV 3CL protease.
In the initial description of DWV, Lanzi et al. identified two potential N-terminal cleavage sites for the 3CL protease with P1 Q
2118 and P1’ G
2119 as well as P1 E
2180 and P1’ G
2181. They also described potential active amino acid residues of the catalytic triad, namely C
2307, H
2190 and D
2225. Recently, the first enzymatic properties have been reported for recombinant DWV’s 3CL protease (position 2119–2409 of the DWV polyprotein), which specifically cleaved the peptide linking the DWV leader protein-VP2 interface [
40]. The N-terminus of DWV 3CL protease, which was already predicted in 2006 [
26], was confirmed experimentally using a bacterial expression system approach [
40,
41]. After the Q
2118 of the potential N-terminal cleavage site was replaced with an alanine, no maturation at the site was observed. The catalytic triad of the DWV 3CL protease was more accurately predicted using a structural model calculated using AlphaFold2 in combination with further sequence alignments. Alanine substitution experiments indicated that the identity of amino acids H
2170, C
2307, and N
2227 is pivotal for enzyme activity, while D
2225 is of minor importance. Another study used a very similar system with a short recombinant DWV 3CL protease fragment and a fluorogenic substrate to further determine the optimum temperature of the enzyme [
41].
Here, we used an E. coli-based expression system to identify the mature products of the processing between the 3CL protease and the 3D-like polymerase (3DL) of DWV. We also generated monoclonal antibodies against the 3CL protease and 3DL to identify the mature proteins and precursors in DWV-infected honey bee pupae. These observations were verified using our molecular clone of DWV-A, demonstrating that the 3CL protease protease activity and the identity of the N-terminal and C-terminal cleavage sites is pivotal for DWV replication in vivo.
2. Materials and Methods
2.1. Viruses and Honey Bees
Honey bee pupae (
Apis mellifera carnica) were collected from the apiary of the Institute of Virology, Justus-Liebig University (Giessen, Hesse, Germany) with the permission of the university. Worker bee pupae of the same age were taken from a single comb, which had been freshly introduced in the colony and subsequently labelled after egg deposition to calculate the average age of pupae. Using sterile tweezers, we extracted 13- to 15-day-old pupae with purple to blue eyes from capped brood cells, transferred them into individual wells of 24-well tissue culture plates, and incubated them at 33 °C in a humidified incubator. Damaged or dead individuals were sorted out the next day to achieve uniform experimental conditions. Virus stocks and mock control bees were checked for contamination from other viruses as described previously [
42].
Our molecular clone of the DWV-A strain 1414 (GenBank KU847397)) and the reverse genetics system was presented earlier [
16]. Briefly, the plasmid DNA of the molecular clone was prepared (Genopure Plasmid Midi Kit; Roche, Basel, Switzerland) and linearized with the restriction enzyme NotI (NEB, Ipswich, MA, USA). Cleavage yielded linear DNA with an SP6 polymerase promoter upstream of the genomic DWV cDNA, which had been cut just downstream of the poly-A tail sequence. Subsequently, the linearized plasmid DNA was purified (Monarch PCR & DNA Cleanup Kit; NEB) and quantified to serve as a template for in vitro RNA transcription (HiScribe SP6 RNA Synthesis Kit; NEB). The RNA was also purified, quantified, and diluted to a final concentration of 1 µg/µL. The bee pupae were transfected with 1 µg of the synthetic RNA by injecting the RNA solution into the thorax segment. Pupae from the same comb were injected with 1 µL of phosphate-buffered saline solution (PBS) to serve as mock controls. All pupae were incubated for 3 days at 33 °C. After the incubation, the bee pupae were frozen at −20 °C, thawed, and then homogenized in 500 µL PBS containing 0.5% Trition X-100. Uniform homogenization was achieved by shaking three times at 4000 rpm for 1 min (MagNA Lyser; Roche, Basel, Switzerland) with the help of stainless-steel beads (Qiagen, Hilden, Germany).
2.2. SDS-PAGE, Western Blotting, and Edman Degradation
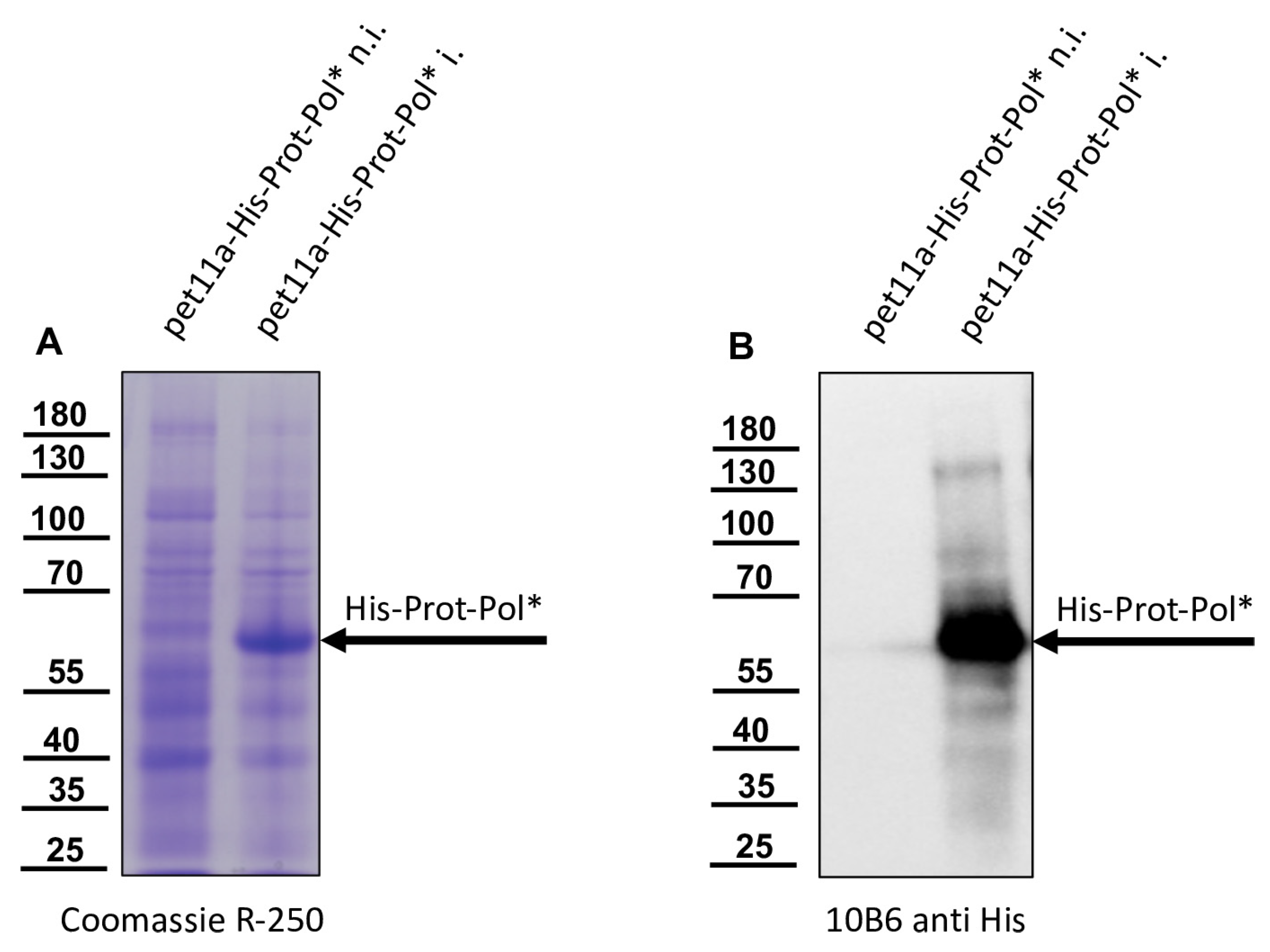
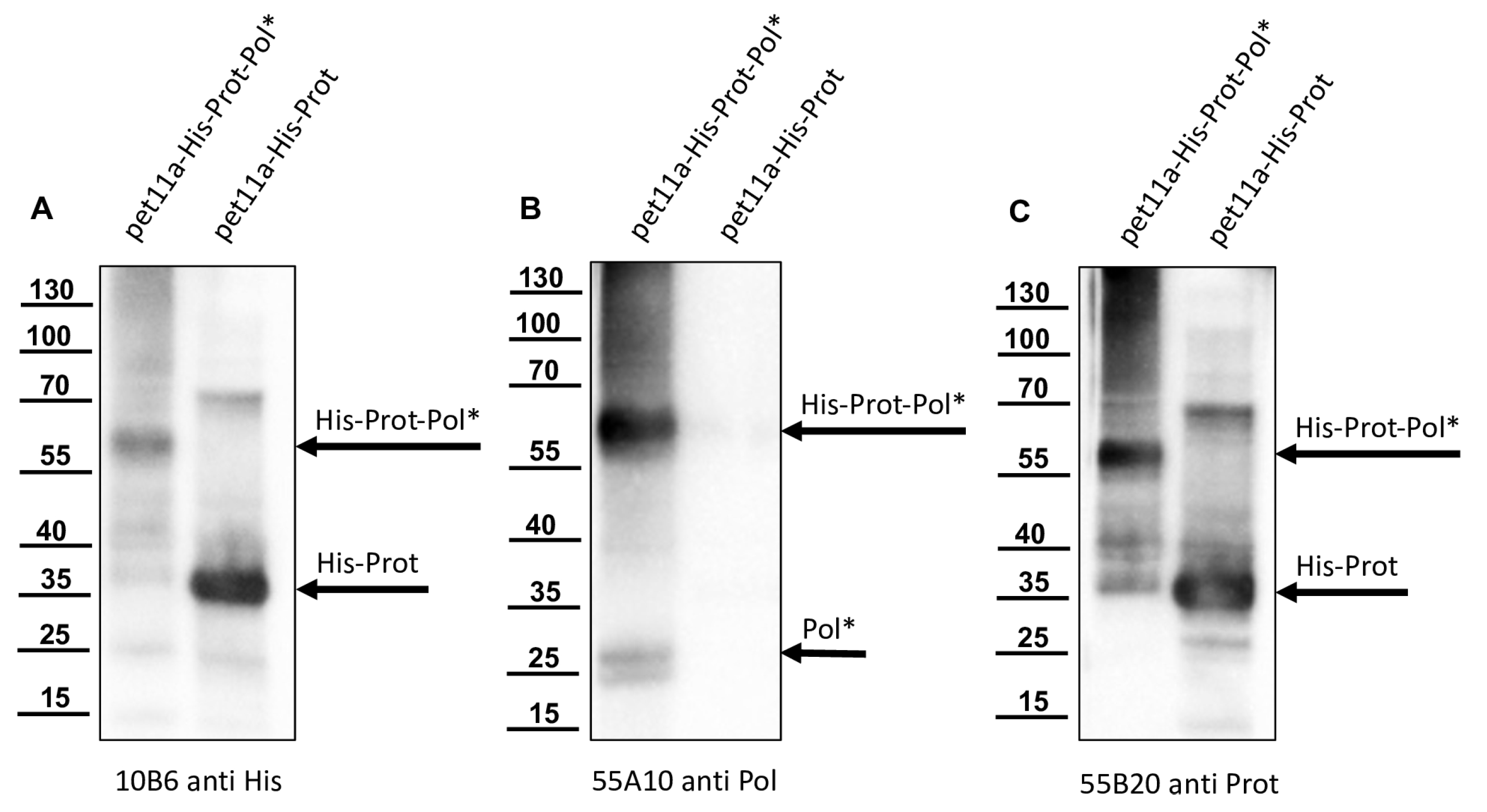
For sodium dodecyl sulfate—polyacrylamide gel electrophoresis (SDS-PAGE), an SDS-containing 6x loading buffer was added to the honey bee pupae lysates, bacterial lysates, or purified proteins. The SDS-PAGE samples were boiled at 95 °C for 5 min and separated in 7.5% polyacrylamide tricine gels. For the analyses, bee pupae were homogenized in the same way, and equal volumes of virus-positive and negative samples were loaded. Although we do not have a Western blot loading control antibody for honey bees, the similar loading of the lanes can be at least partially reconstructed on the basis of similarly strong background bands. The gels were either stained with Coomassie brilliant blue R-250 or transferred onto Western blot membranes (Pall, Pensacola, FL, USA).
Nitrocellulose membranes were blocked in 5% skim milk (Carl Roth, Karlsruhe, Germany) PBS with 0.1% Tween-20 (Invitrogen, Karlsruhe, Germany). Primary murine monoclonal antibodies (Mabs) were used for protein detection as indicated. Antibody binding was visualized using secondary goat-anti-mouse HRPO conjugates (Dianova, Hamburg, Germany) together with ECL-Prime (GE Healthcare, Chicago, IL, USA). Photon emission was recorded and visualized using a digital imaging system (ChemiDoc; Bio-Rad, Feldkirchen, Germany). Mabs against the DWV 3CL protease (Mab 55B20) and 3DL polymerase (Mab 55A10) were generated in this study. Mab VP1A1 against DWV VP1 and Mab 10B6 anti-His were established earlier [
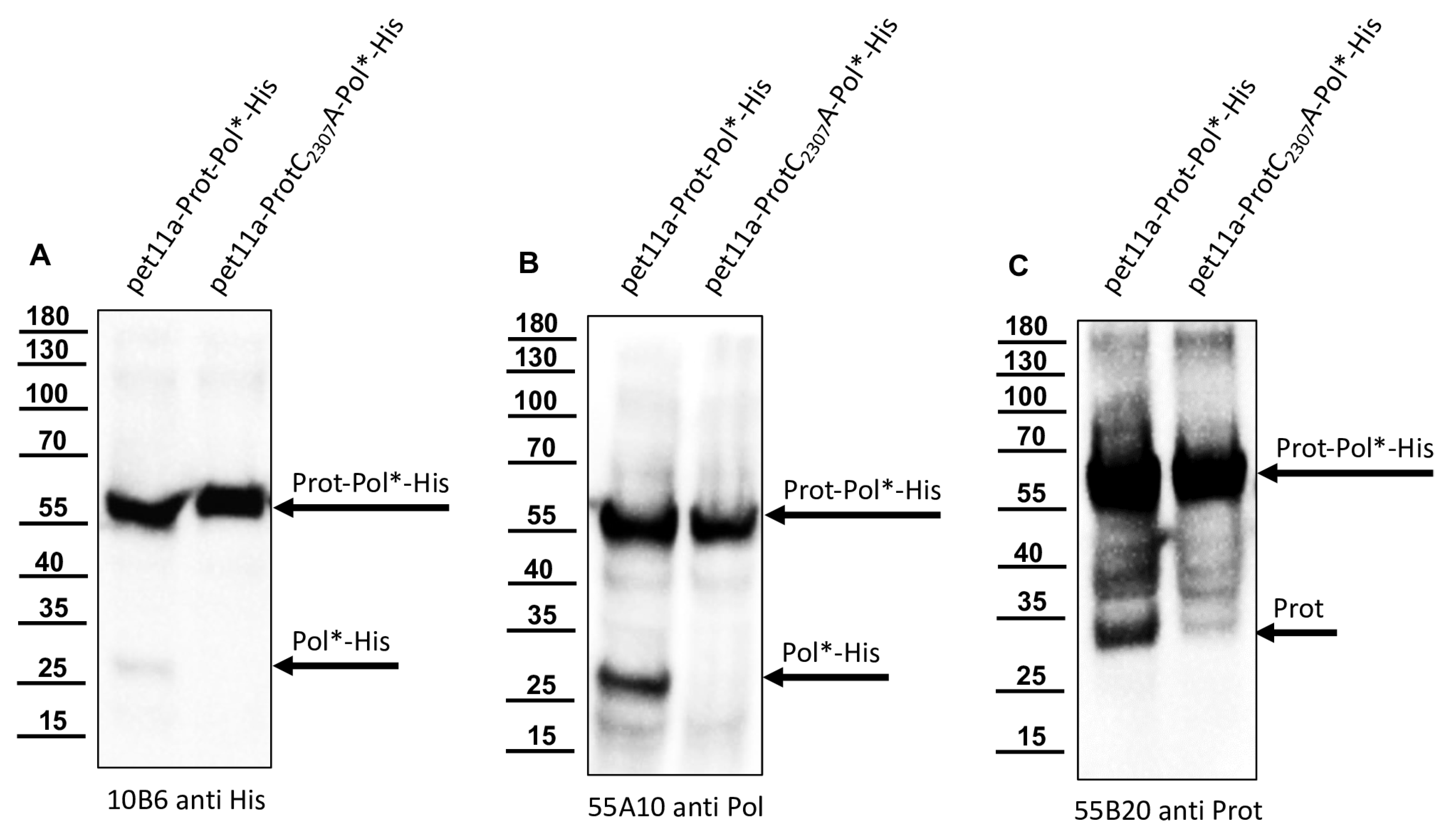
16]. The anti-Flag Mab M2 was commercially purchased (Merck, Darmstadt, Germany).
N-terminal protein sequencing was carried out via phenyl isothiocyanate chemistry as developed by Pehr Edman and analyzed using high-performance liquid chromatography [
43]. For this purpose, proteins were transferred to polyvinylidene fluoride (PVDF) membranes using borate buffers, stained with Ponceau S solution (Carl Roth), and excised with a razor blade. Automated Edman degradation was performed by a commercial supplier (Proteome factory, Berlin, Germany).
2.3. Recombinant Protein Production
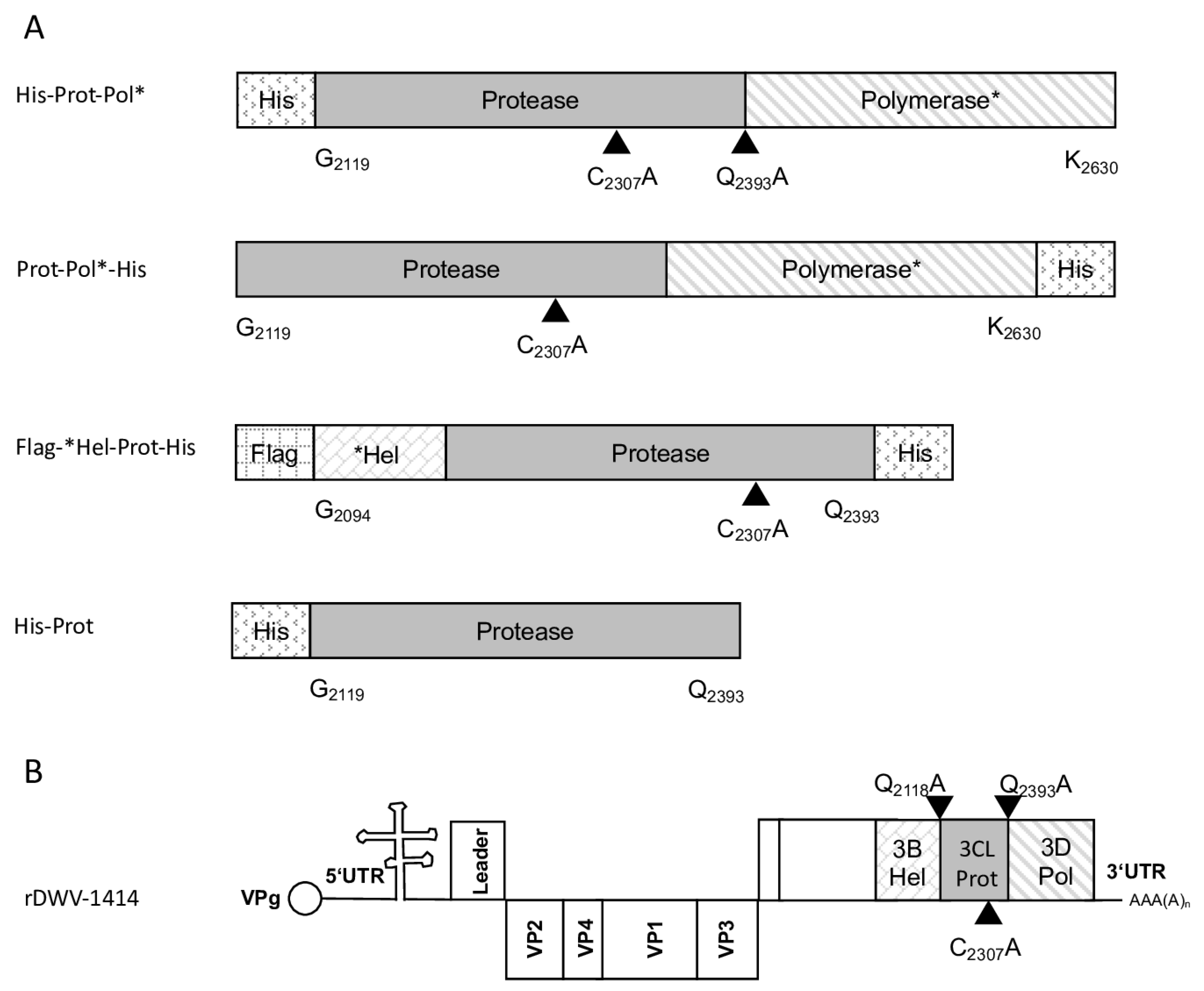
The molecular clone of the DWV-A strain 1414 served as a DNA template to amplify the 3CL protease and parts of the 3DL gene. The ORF region encoding amino acids G
2119-K
2630 was amplified via PCR (Phusion High-Fidelity DNA Polymerase; NEB) using oligonucleotides 5′-3C-Prot_fwd and Internal-Pol_rev. The sequences of the oligonucleotides used in this study are listed in
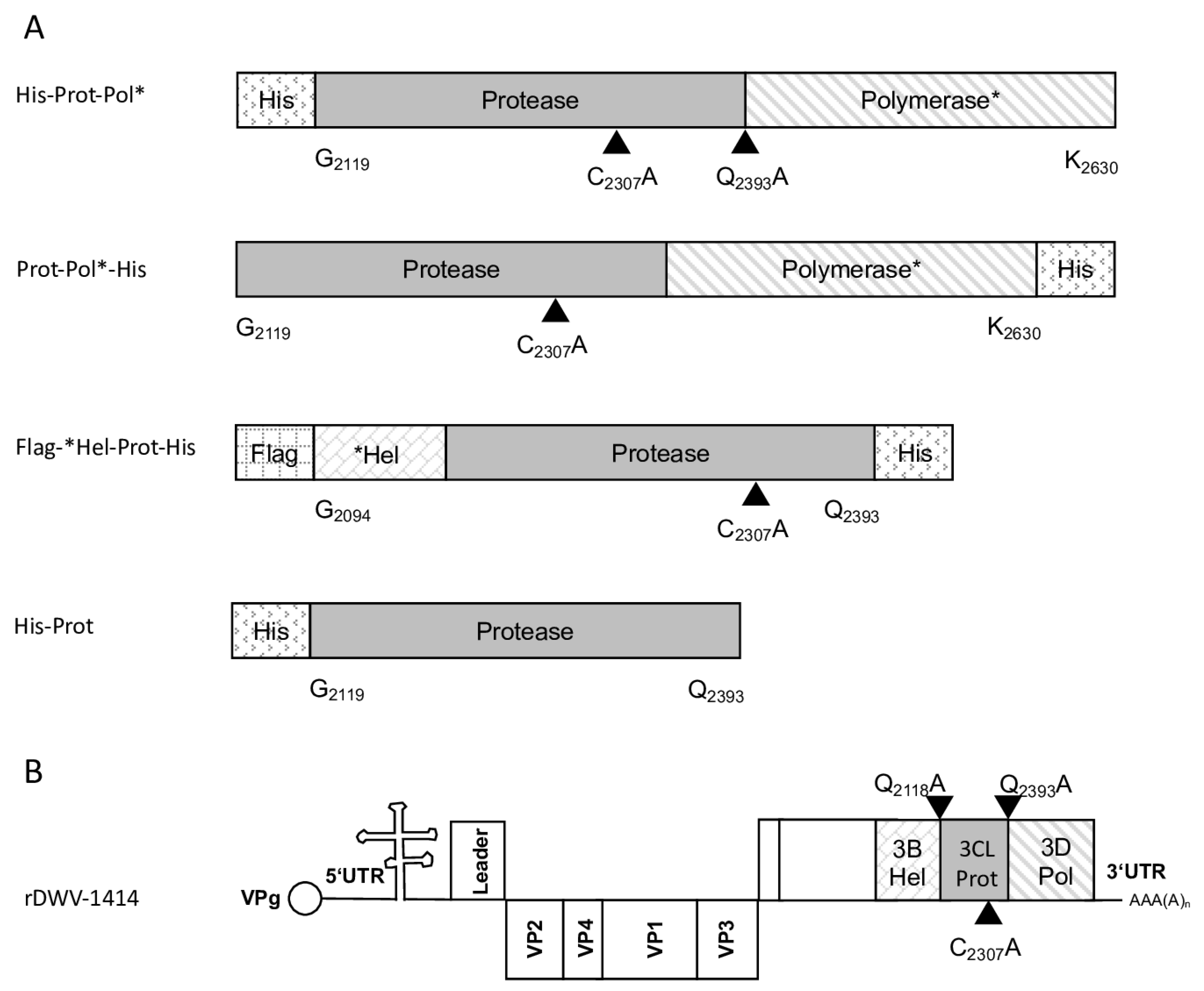
Table 1. A suitable vector backbone of a bacterial expression plasmid (pet11a; Merck KGaA, Darmstadt, Germany) containing an N-terminal 10x-His-tag was amplified using the oligonucleotides 5′-3C-overhang-pet11a_rev and Internal-Pol-overhang-pet11a_fwd, adding complementary extensions for DNA recombination. After purification of the respective PCR products, an in vitro DNA assembly reaction (NeBuilder; NEB) yielded the expression vector pet11a-His-Prot-Pol*. The recombinant expression constructs used in this study and a genome scheme of DWV mutations are presented in an overview figure (
Figure 1).
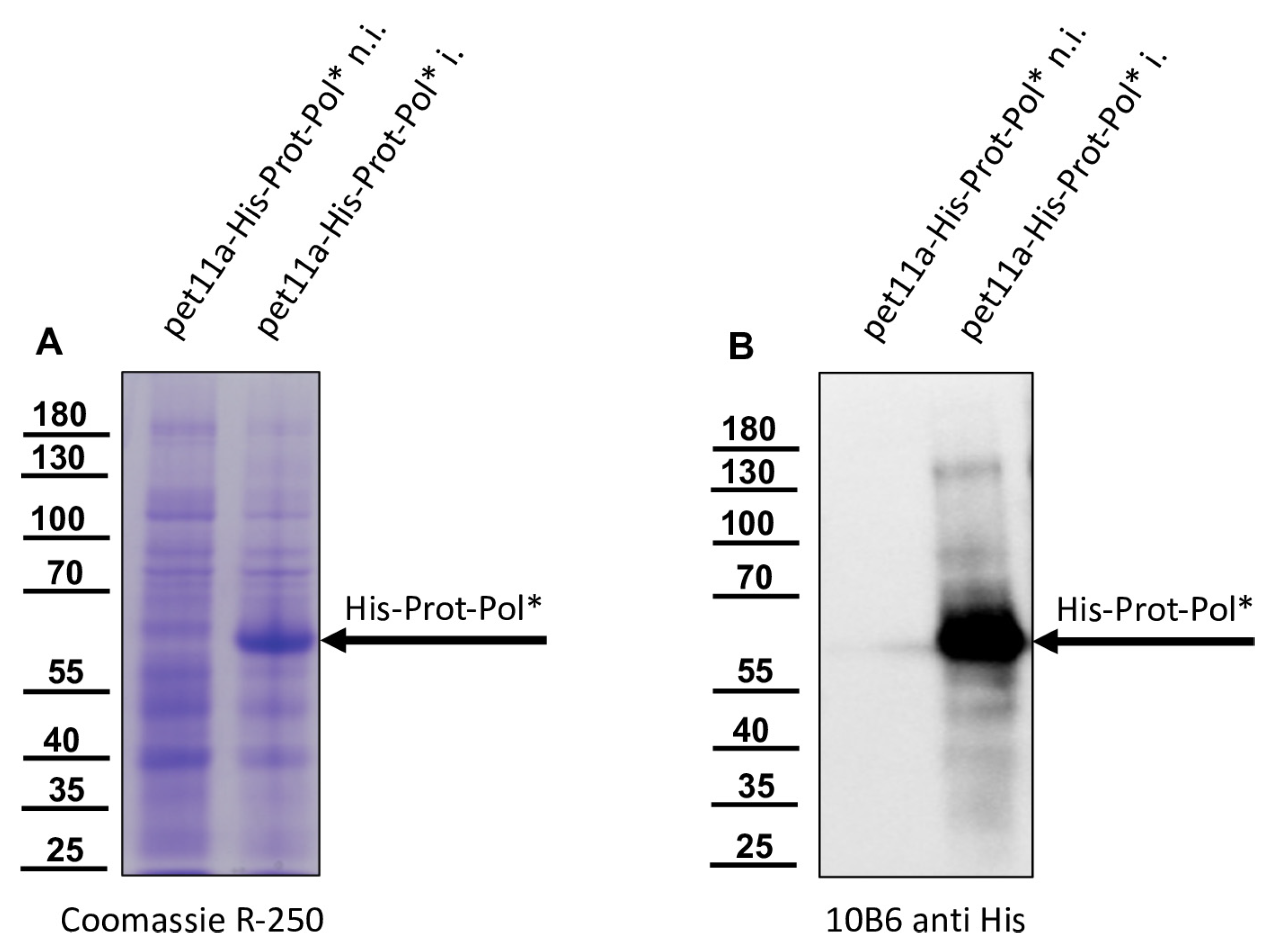
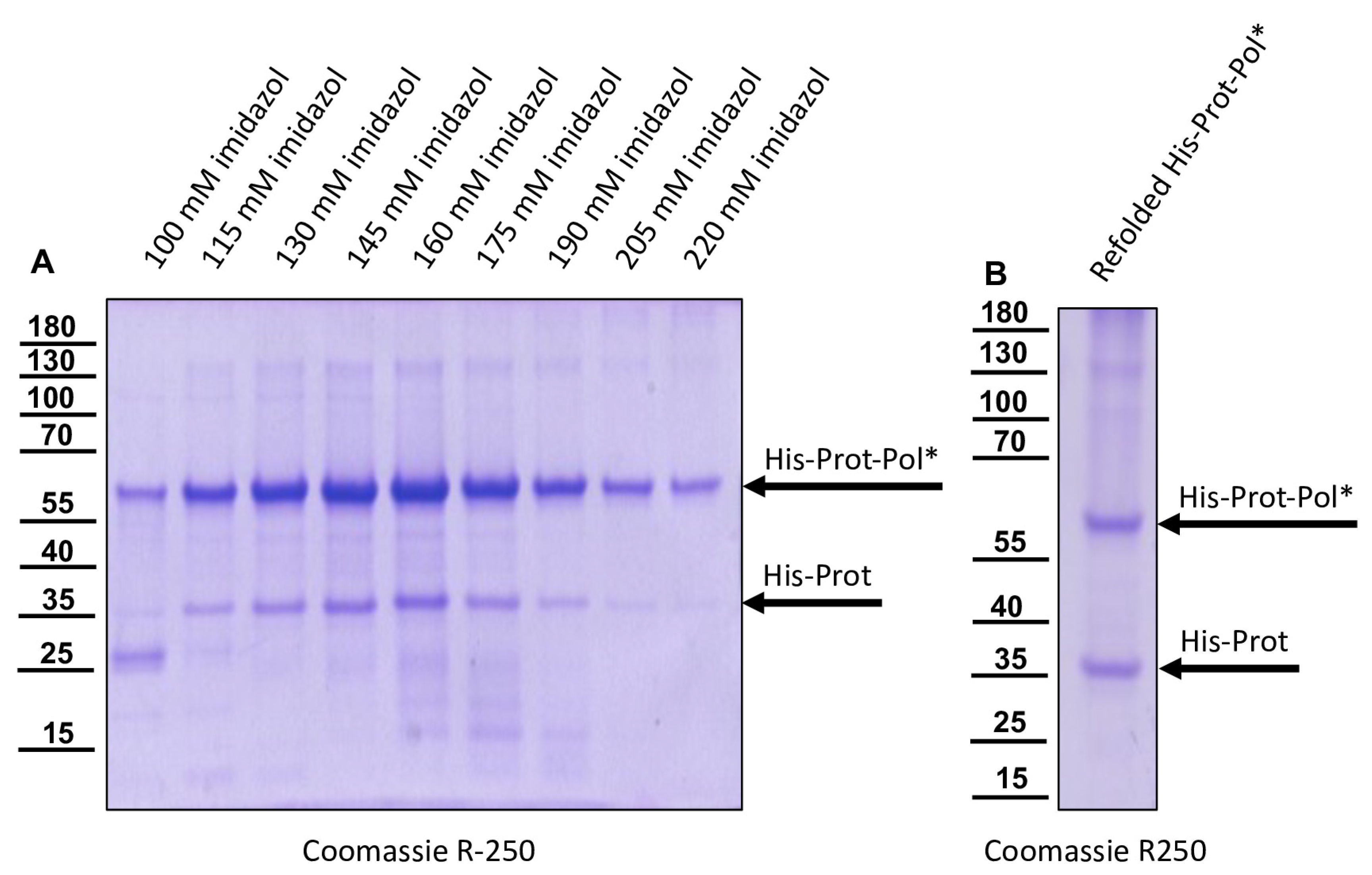
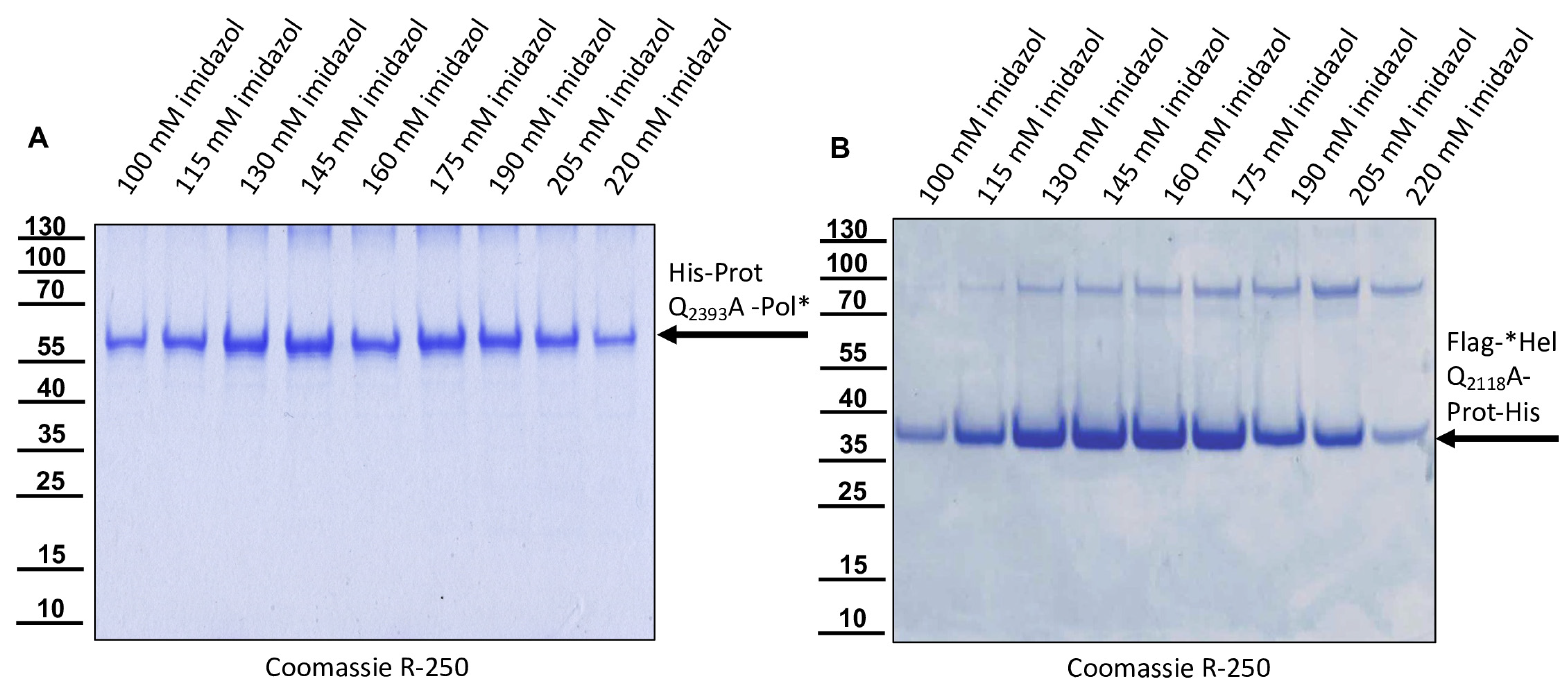
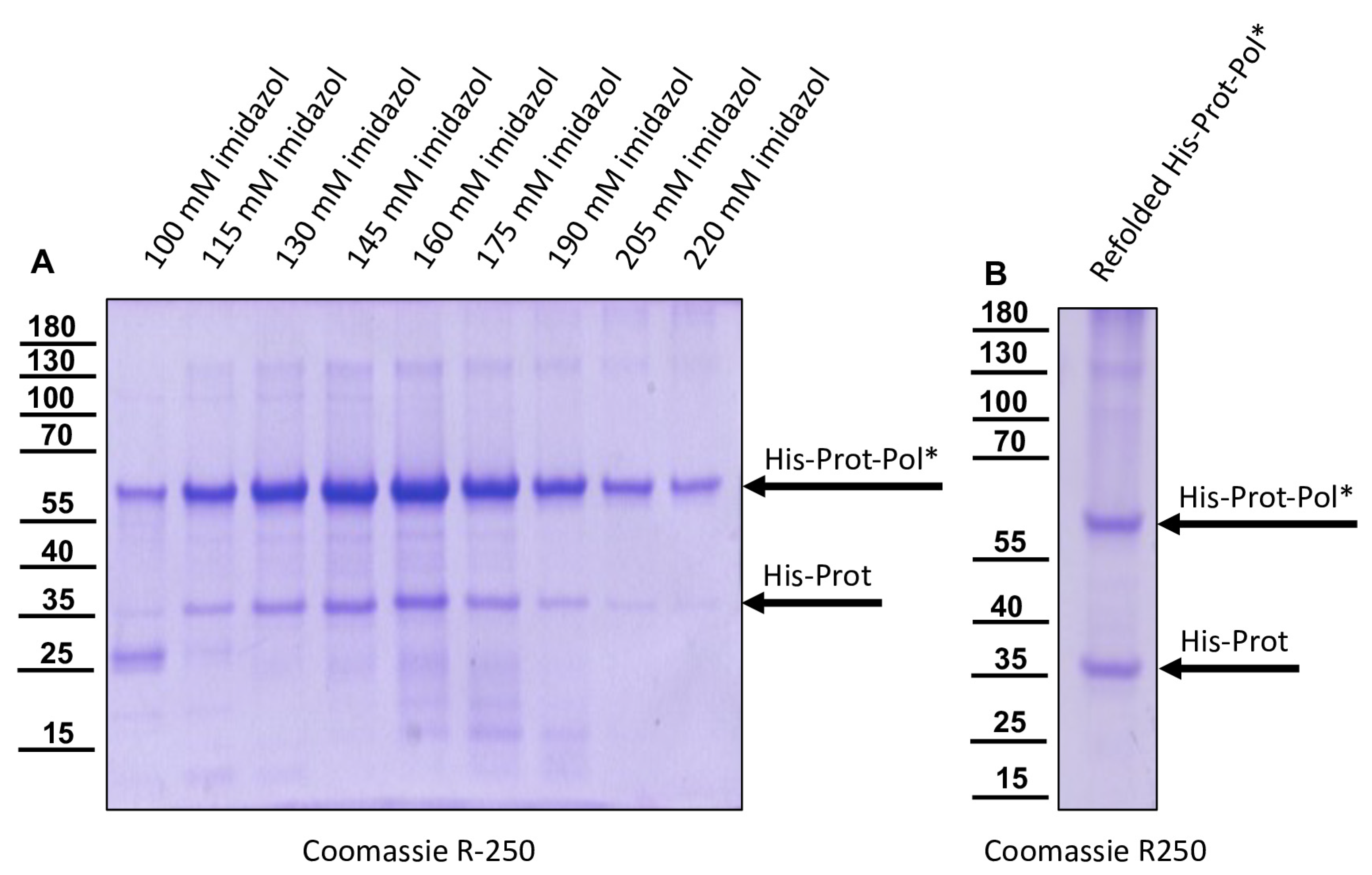
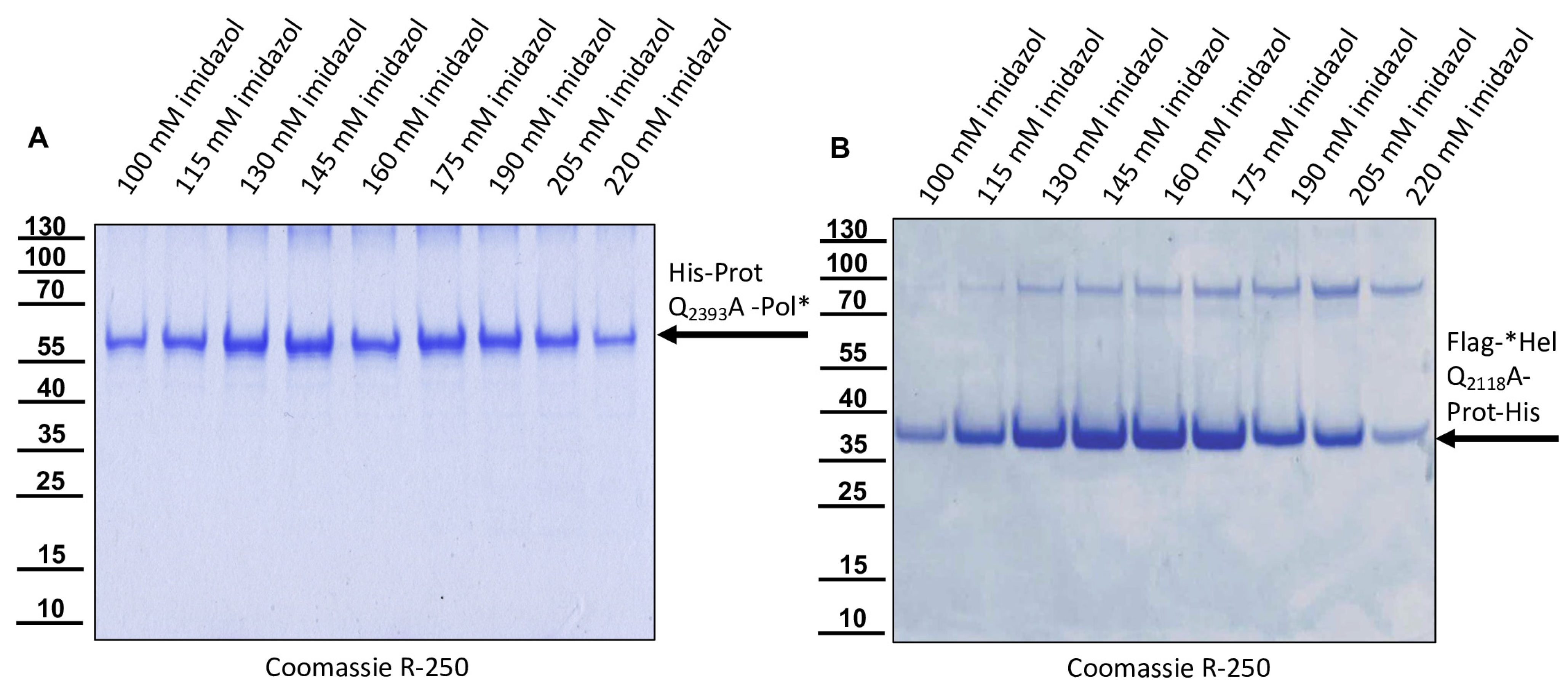
The DNA cloning procedure was carried out in non-expression E. coli host cells of strain HB101. For expression, purified plasmid DNA was transferred into expression host cells of the E. coli strain Rosetta (Merck) containing a chromosomal copy of the T7 RNA polymerase gene under lacUV5 control. Rosetta cells were grown in Luria–Bertani media until they reached an optical density at 600 nm (OD600) of 0.7–0.8. Then, IPTG was added to a final concentration of 1 mM to induce expression of T7 RNA polymerase, which in turn transcribed the recombinant protein gene. After an incubation of 4 h at 37 °C, the cells were harvested via centrifugation (4000× g for 10 min), and the cell pellet was resuspended in PBS containing 0.5% Triton X-100. Cell lysis was performed using three cycles of freezing and thawing according to the manufacturer’s recommendation. The His-Prot-Pol* protein was completely insoluble after cell lysis—most probably due to inclusion body formation. Hence, the lysate was cleared via centrifugation (10,000× g at 4 °C for 30 min), the supernatant was discarded, and the remaining pellet of insoluble material was resolubilized in PBS containing 8 M urea. The resolubilized protein solution was subjected to immobilized metal ion affinity chromatography (IMAC) on an ÄKTA pure 25 device (Cytiva, Marlborough, MA, USA). After binding to Ni-Sepharose (HisTrap HP; Cytiva) and being washed with 8 M urea PBS, the protein was eluted in 8 M urea PBS, using an imidazole gradient (0 to 500 mM). The elution process was monitored via UV absorption (280 nm), and the identity of the eluted protein was verified using SDS-PAGE and subsequent Western blot analysis with Mab 10B6 anti-His. The protein-containing elution fractions were pooled, and urea was diluted out stepwise. The addition of PBS to the denatured samples was sufficient to fold the protein back to a soluble state without recognizable losses. The soluble protein was re-concentrated via ultrafiltration (Amicon Ultra-15, 30 kDa MWCO; Millipore, Burlington, MA, USA) and washed twice to remove last urea contaminants.
2.4. Generation of Monoclonal Antibodies
The recombinant His-Prot-Pol* was homogenous and suitable for the immunization of mice, which was carried out by a commercial provider (Davids Biotechnologie GmbH, Regensburg, Germany). After successful immunization of three Balb/c mice, spleen cells of hyperimmune animals were harvested, fused with SP2/0-Ag14 myeloma cells, and selected using the hypoxanthine-aminopterin-thymidine (HAT) method. The supernatants of individual hybridoma cell clones were screened for reactive antibodies via indirect enzyme-linked immunosorbent assay (ELISA) again using the recombinant His-Prot-Pol* as an antigen source. For primary validation of Mab reactivity, we harvested cells of induced and non-induced bacterial cultures expressing His-Prot-Pol*. These cells were lysed, subjected to SDS-PAGE, and blotted onto nitrocellulose membranes. Subsequently, the supernatants of all ELISA-reactive hybridoma clones (more than 100) were tested. To determine whether the individual antibodies were directed against the protease or against the polymerase moiety of our immunogen, another expression plasmid was generated. This plasmid contained only the 3CL protease (G2119-Q2393) as experimentally determined via Edman degradation and described in the results section. The expression plasmid pet11a-His-Prot was generated by deleting the polymerase sequences from the expression plasmid pet11a-His-Prot-Pol* using a Phusion reaction (NEB) using the oligonucleotides NcoI-3′-3C_rev and NcoI-Stop-pet11a_fwd. The PCR was digested with the enzymes DpnI (NEB), which removed template DNA, and NcoI (NEB), which generated sticky PCR product ends. The product of digestion was purified and ligated using T4-DNA-Ligase (Instant Sticky-end Ligase Master Mix, NEB). Cell lysates from induced Rosetta cultures expressing either His-Prot or His-Prot-Pol* were resolved side-by-side in SDS-PAGE and blotted to analyze to binding of Mabs to the different protein domains.
2.5. Analysis of the Processing of Recombinant DWV Polyprotein Fragments
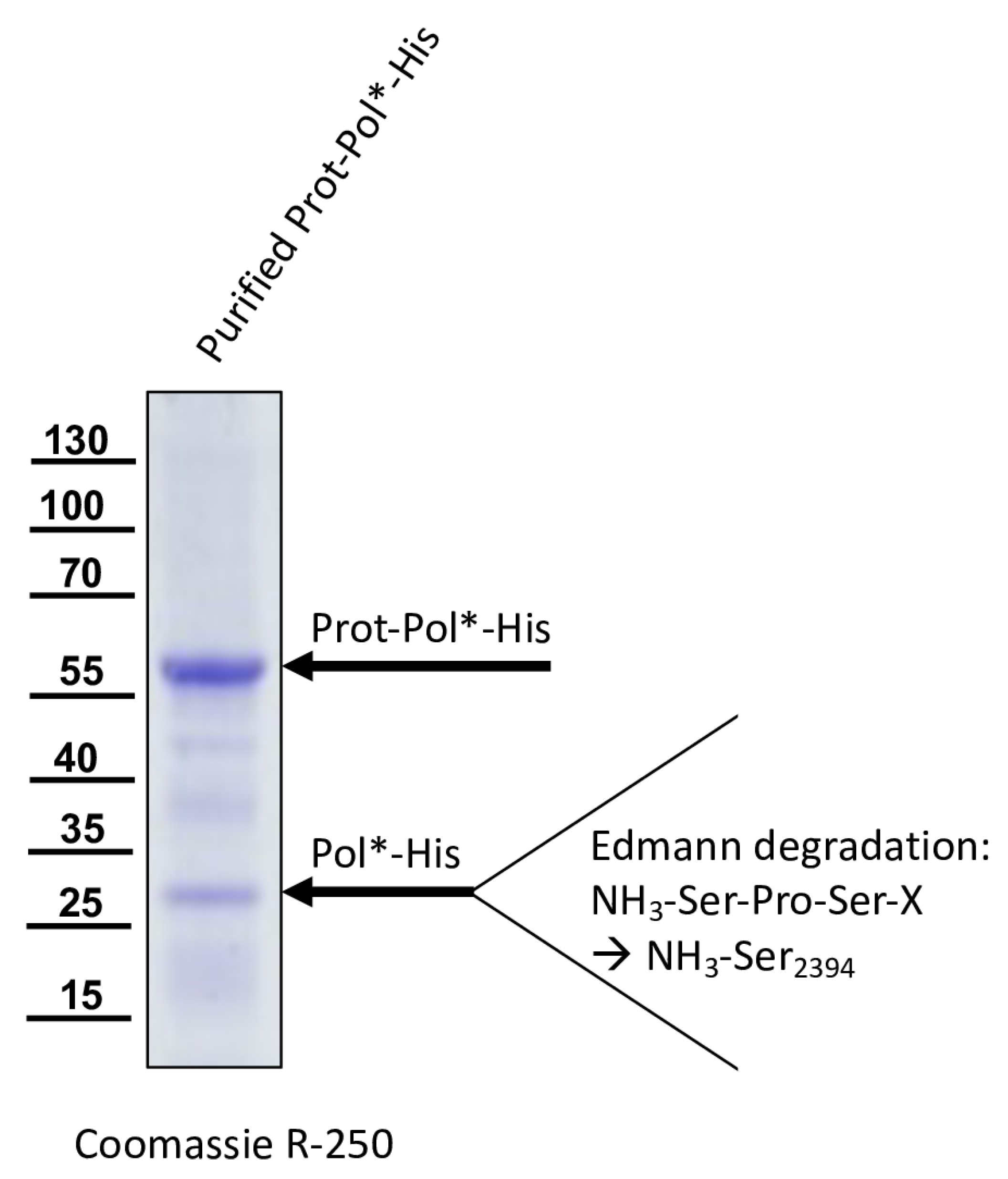
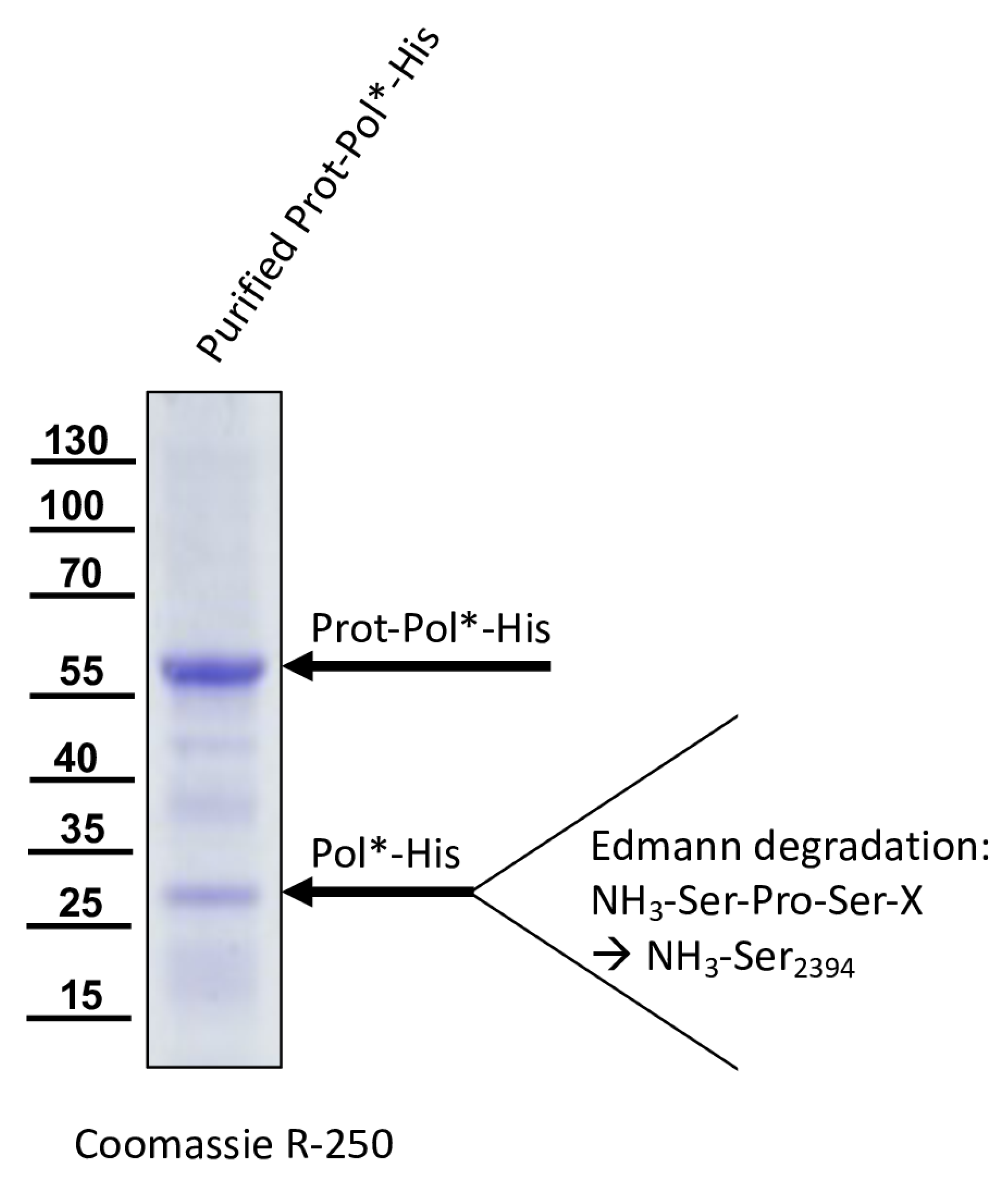
For the determination of the C-terminal cleavage site of the DWV 3CL protease, we C-terminally His-tagged the identical polyprotein fragment consisting of the 3CL protease fused to the stretch of the N-terminus of the polymerase region (Pol*). The pet11a-Prot-Pol*-His vector thus encompassed the polyprotein amino acids G2119-K2630. A PCR product of this ORF fragment was generated using the oligonucleotides pet11a-overhang-5′-3C_fwd and His-overhang-Internal-Pol_rev. A suitable vector backbone with a C-terminal His-tag for the expression plasmid was amplified via PCR, using oligonucleotides Internal-Pol-overhang-His_fwd and 5′-3C-overhang-pet11a_rev. The PCR products were recombined via DNA assembly.
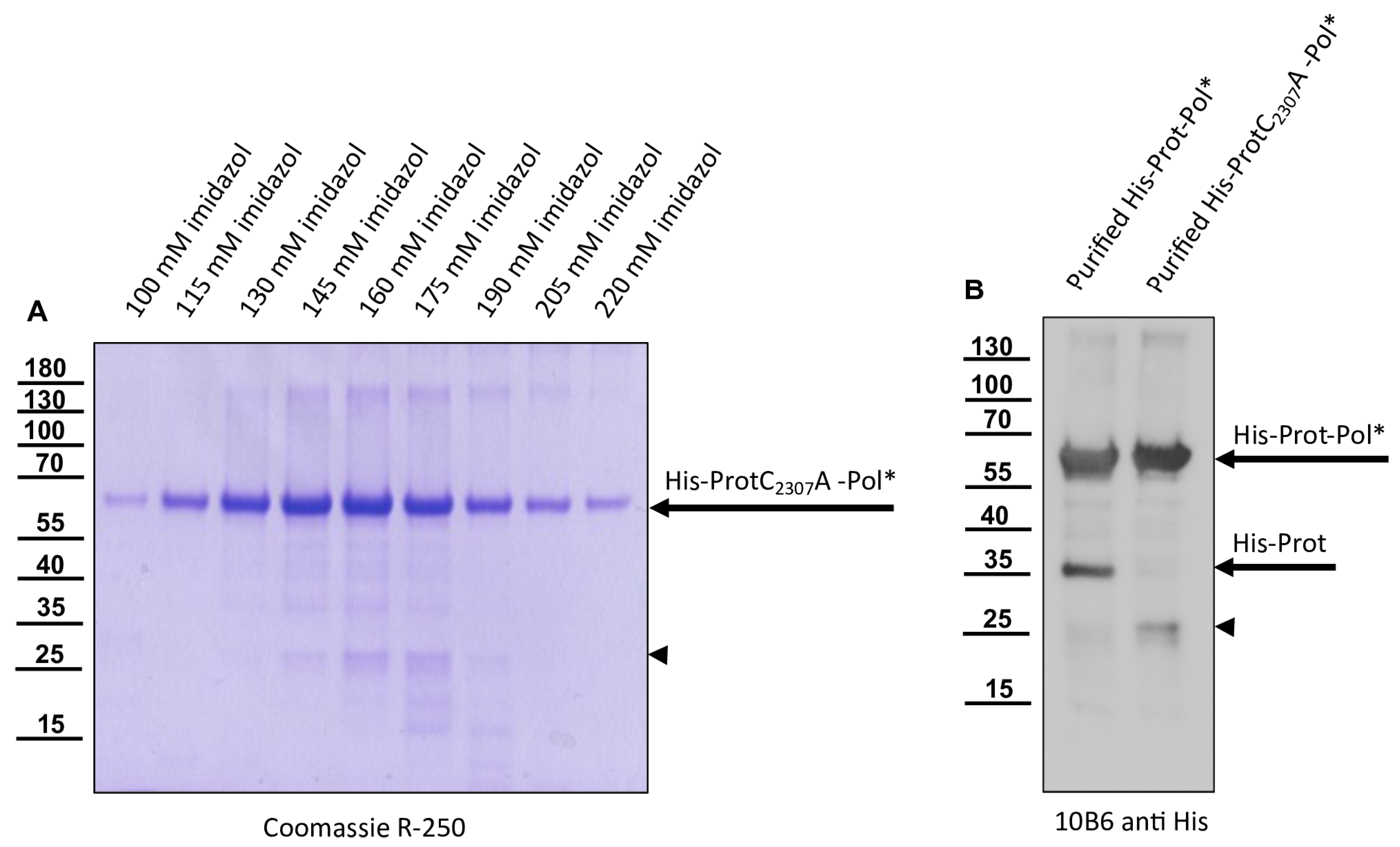
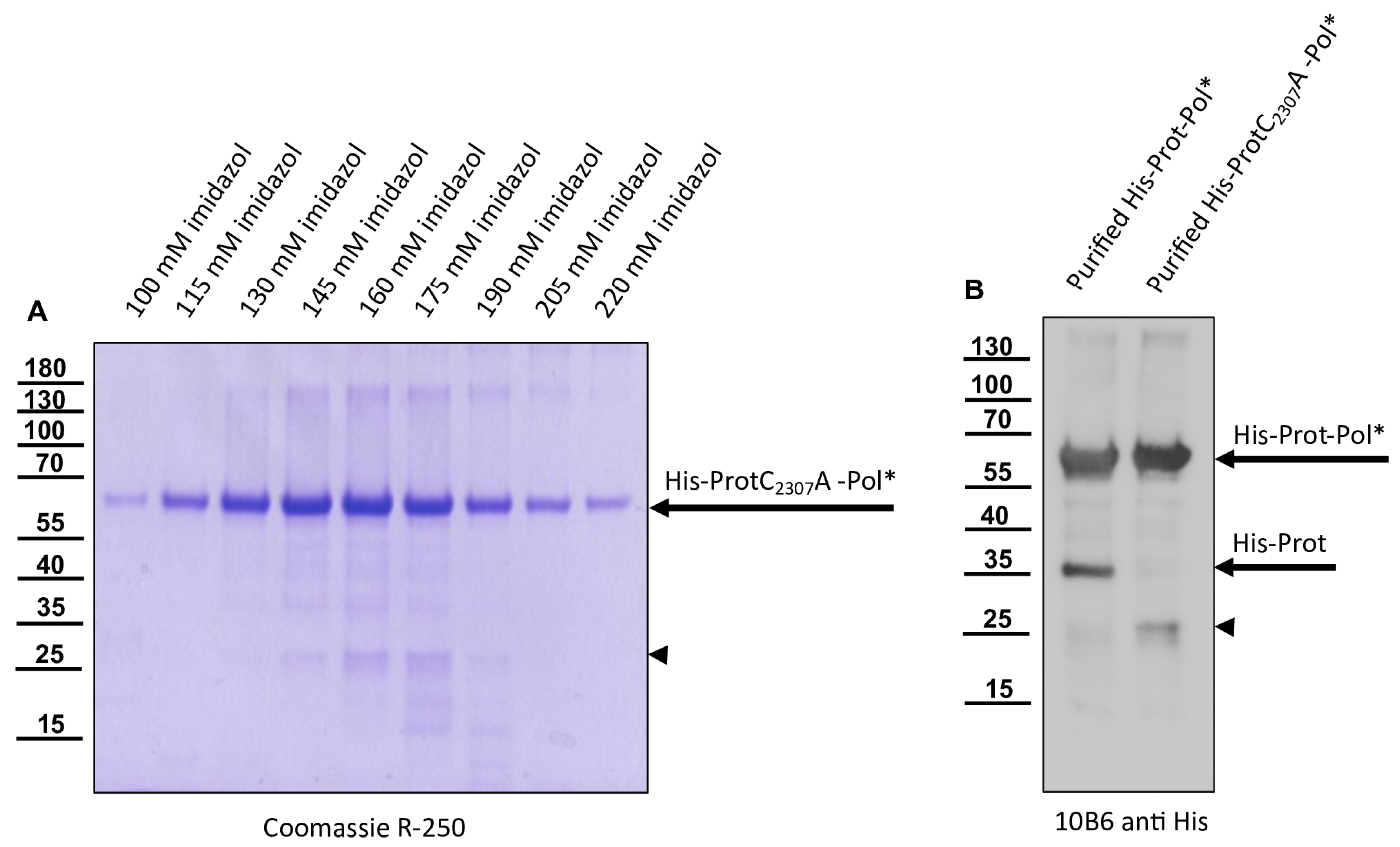
The 3CL protease was inactivated in a negative control plasmid by introducing the amino acid exchange C
2307A. Replacement of the active cysteine within the catalytic triad should inactivate the enzyme as shown by Yuan et al. [
40], and hence block autocatalytic processing. Oligonucleotides 3C-C
2307A_fwd and Internal-Beta-Lactamase_rev were used to amplify one fragment of the plasmid, whereas 3C-C
2307A_rev and Internal-Beta-Lactamase_fwd were used for the other to generate pet11a-ProtC
2307A-Pol*-His.
The glutamine at position 2393 was changed to an alanine (Q2393A) to destroy the determined cleavage site between the protease and the polymerase. For this purpose, two separate fragments were again amplified via PCR, and the mutation was encoded within the primer sequences. Homologous recombination was used to reassemble the fragments using the same strategy with a second assembly site in the beta-lactamase gene, resulting in pet11a-ProtQ2393A-Pol*-His.
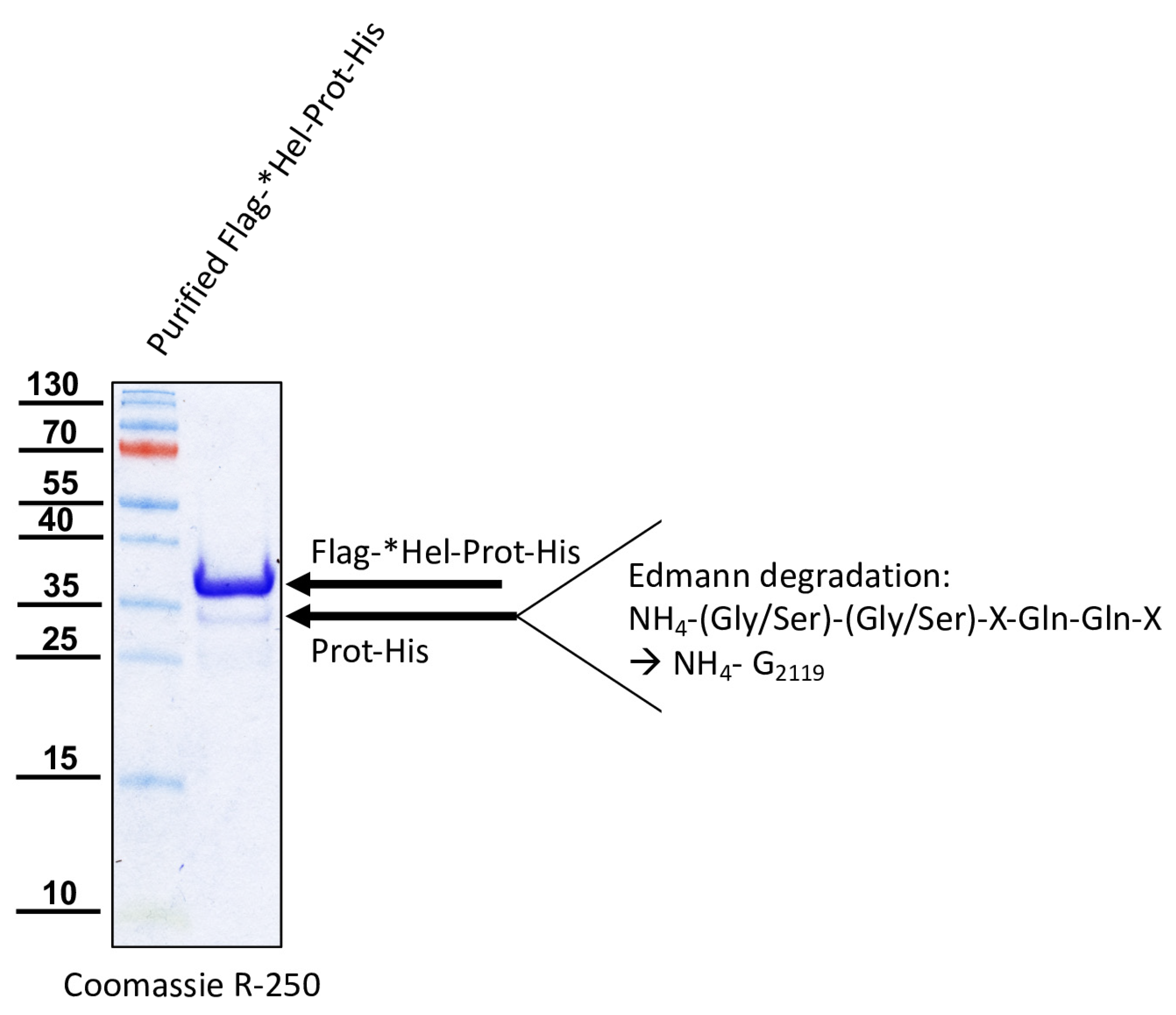
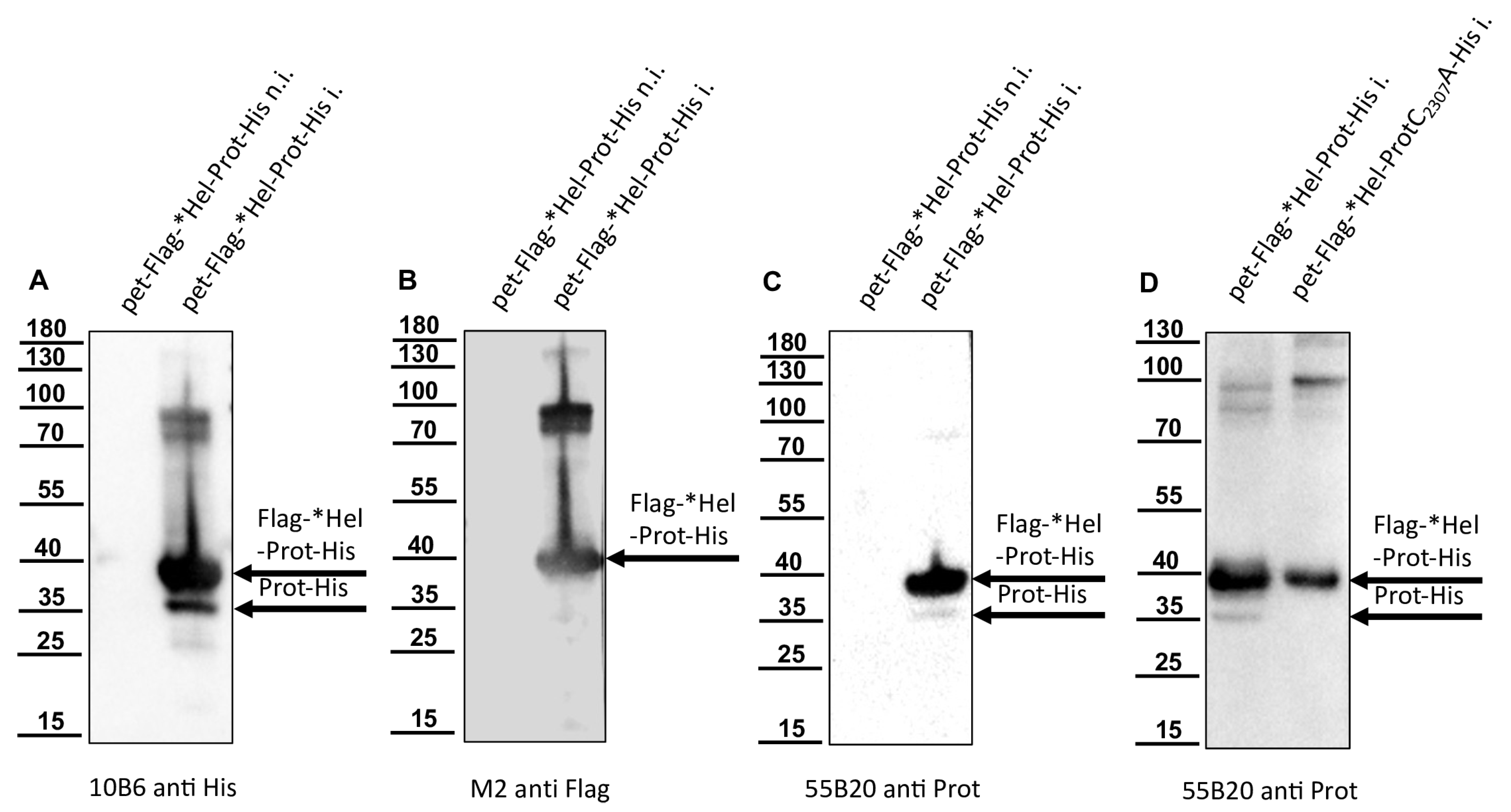
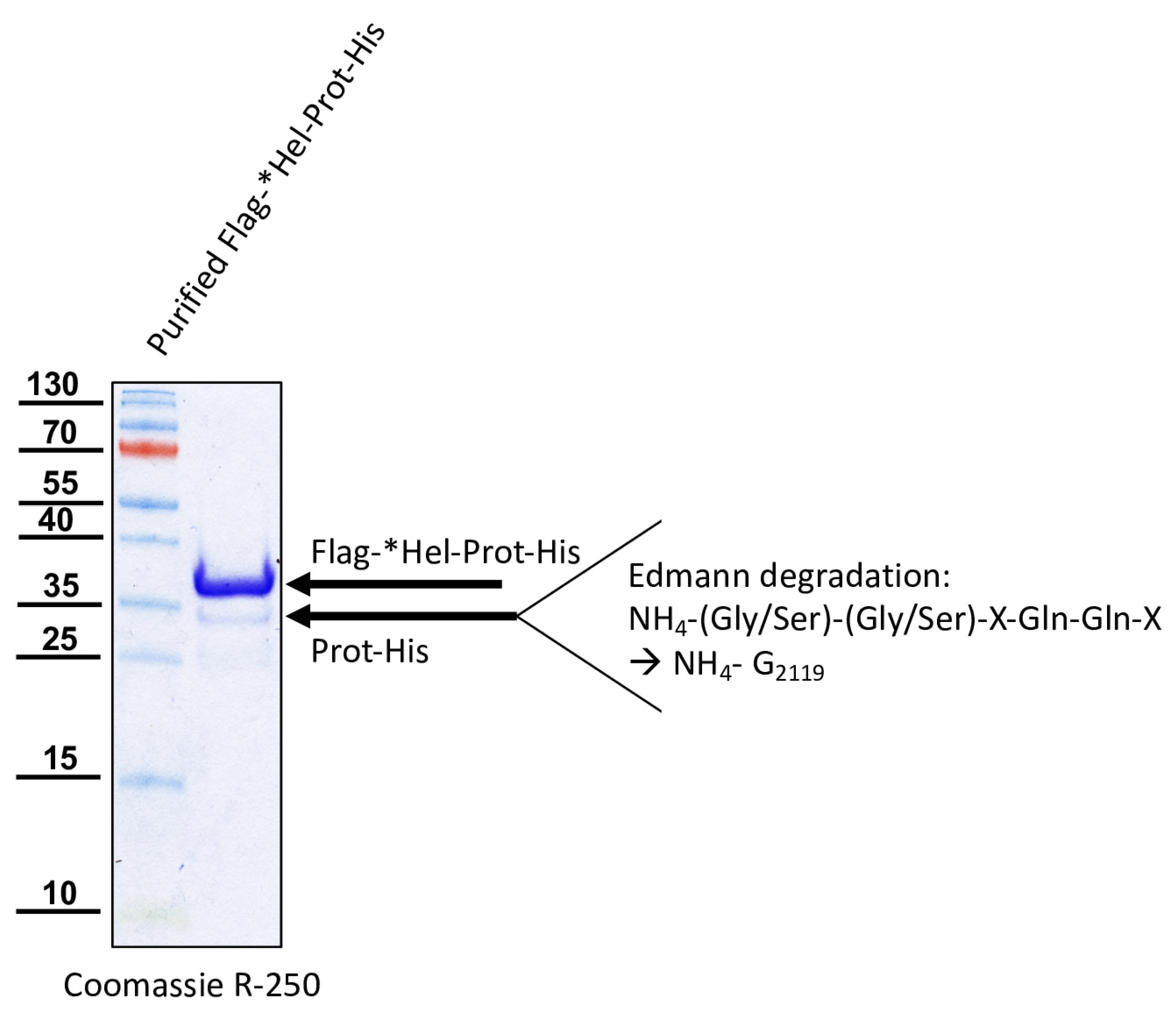
For the determination of the N-terminal cleavage site of the DWV 3CL protease, we generated another N- and C-terminally tagged polyprotein fragment consisting of an N-terminal Flag-tag, a short stretch from the C-terminus of the helicase (*Hel), the complete 3CL protease and a C-terminal His-tag sequence. The DWV ORF sequence coding amino acids G2094–Q2393 was amplified using the oligonucleotides Flag-Internal-Hel_fwd and His-3′-3C_rev adding the Flag-tag sequence on the 5′-end and a His-tag sequence at the 3′-end of the PCR product. A suitable backbone of a pet11a expression vector, already containing an N-terminal Flag-tag and a C-terminal 10x His-tag was amplified via PCR using the oligonucleotides ATG-Flag _rev and 3C-overhang-His-tag_fwd. The in vitro assembly reaction generated the expression vector pet11a-Flag-*Hel-Prot-His.
A negative control with the mutation C2307A was also generated. For this purpose, we used the oligonucleotides 3C-C2307A_fwd and 3C-C2307A_rev and again employed the beta-lactamase gene assembly site. The respective expression construct was termed pet11a-Flag-*Hel-ProtC2307A-His.
The glutamine at position 2118 was changed to an alanine (Q2118A) to destroy the determined cleavage site between the helicase and the protease. For this purpose, two separate fragments were again amplified via PCR and the mutation was encoded within the primer sequences (3C-Q2118A_fwd and 3C-Q2118A_rev). Homologous recombination was used to reassemble the fragments using the same strategy with a second assembly site in the beta-lactamase gene, resulting in pet11a- Flag-*HelQ2118A-Prot-His.
2.6. Analysis DWV Polyprotein Processing In Vivo
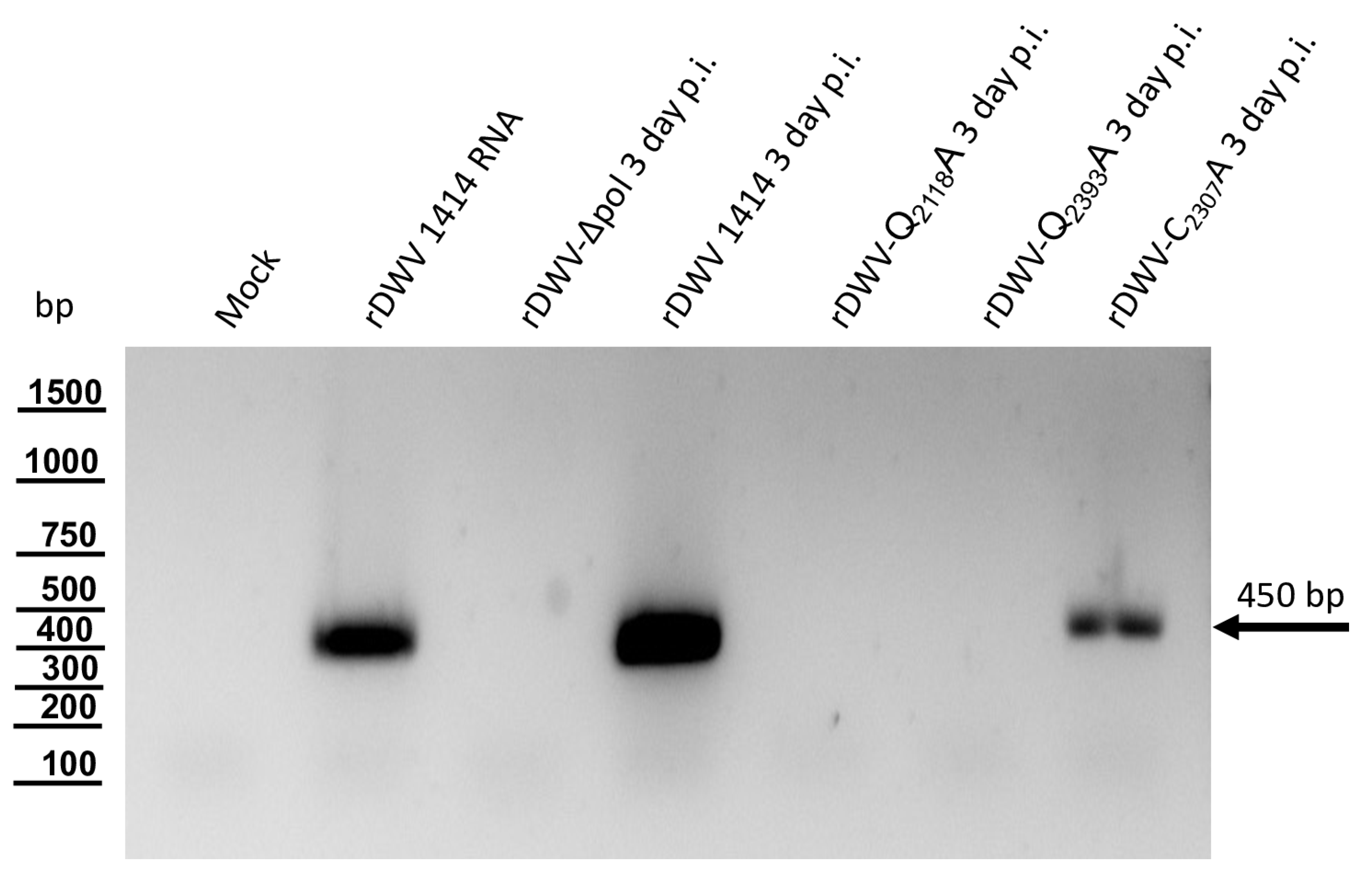
DNA assembly reactions were used to introduce single amino acid exchanges into the ORF of our molecular clone of DWV using a very similar strategy. The cysteine at position 2307 was exchanged against alanine employing the same set of oligonucleotides using the assembly sites at the target position as well as in the beta-lactamase gene. Only the PCR products became significantly larger because the entire viral genome and the pBR322 vector framework had to be amplified. An assembly of the respective PCR products yielded a plasmid harboring the mutated genome of rDWV-C2307A. In a subsequent attempt, the glutamine at position 2118 was exchanged against alanine using oligonucleotides 3C-Q2118A_fwd and 3C-Q2118A_rev to validate the importance of the cleavage site in the viral infection cycle using clone rDWV-Q2118A. Similarly, Q2393 was exchanged against an alanine using the oligonucleotides 3C-Q2393A_fwd and 3C-Q2393A_rev. Consequently, this cleavage site mutant cDNA clone was termed rDWV-Q2393A. The histidines at positions 2170 and 2190 were exchanged against alanine employing oligonucleotides 3C-H2170A_fwd/_rev and 3C-H2190A_fwd/_rev. The aspartates at positions 2199 and 2225 were exchanged against alanine employing oligonucleotides 3C-D2199A_fwd/_rev and 3C-D2225A_fwd/_rev. And finally, the asparagine at position 2227 was exchanged against an alanine with the oligonucleotides 3C-N2227A_fwd/_rev. The resulting virus were named in a consistent scheme as rDWV-XnnnnA.
The successful mutagenesis and integrity of the plasmids generated in this study were verified restriction enzyme mapping, initial Sanger sequencing (Microsynth, Balgach, Switzerland), and complete ONT plasmid sequencing (Full PlasmidSeq, Microsynth), which documented error-free cDNA clone sequence. Further details on the DNA cloning and recombination strategies used here will be provided upon request.
4. Discussion
The individual virus species within the order
Picornavirales are very diverse and grouped in eight separate families. While members of the families
Caliciviridae,
Picornaviridae,
Secoviridae,
Polycipiviridae,
Solinviviridae, and
Marnaviridae infect vertebrates, plants, or algae, the ifla-, dicistro-, polycipi- and solinviviruses (families
Iflaviridae,
Dicistroviridae,
Polycipiviridae, and
Solinviviridae) exclusively infect invertebrate hosts. Despite their different host range and remarkable sequence divergence, the members of the order
Picornavirales exhibit many similarities. Common to them all, for example, is a proteolytically processed polyprotein including a block of replicase enzymes consisting of a superfamily III helicase, a proteinase with a chymotrypsin-like structure and a superfamily I RdRp [
23]. The viral polyprotein undergoes a complex processing during and after IRES-mediated translation. First, larger blocks of functionally related proteins (P1, P2, and P3 block) are released, which subsequently give rise to the individual mature proteins. However, a spectrum of different types of processing is observed in the different families and genera, which are related to the different translational strategies and differences in the genome structures [
44].
A well-studied and typical example for these processes is the enterovirus polyprotein. In enteroviruses, a viral autoprotease (2A) splits off the P1 block already in translation due to ribosomal skipping of the peptide bond releasing the structural protein region from the nascent remaining polyprotein (P2 and P3) [
44]. Subsequent or secondary cleavages are mediated by the activity of the main protease (3C) cleaving between the mature viral structural proteins, except for the VP0 cleavage (between VP2 and VP4). In the non-structural proteins, a complex series of events starts with the release of the 3C protease or, more precisely, with the release of its precursors (3CD) via cis-acting auto-proteolysis at the 3B-3C bond. Further trans-cleavage sites at the 2B-2C and 2C-3A bonds are highly sensitive to 3C-mediated catalysis and are efficiently cut. Therefore, an intact P2-P3 block precursor is rarely seen. In contrast, the rather stable 2BC from the P2 block and the 3AB and 3CD precursors from the P3 block are only inefficiently converted into the mature proteins, so that these precursors dominate in infected cells. At least in the case of cleavage of the 3C-3D bond, it can be assumed that cleavages occur mainly as autocatalytic cis-events, since no dilution effects on cleavage have been observed in recombinant purified 3CD. The 3A-3B bond is very stable and is likely to be cleaved only following specific protein–RNA interactions that lead to covalent binding of the 3B to the 5’-end of the genome (viral protein genome-linked; VPg). For the protease, basically four 3C-containing protein species (intact P3 block, 3ABC, 3CD, and mature 3C) can be found in the infected cells. However, 3CD and mature 3C are the most abundant and the other proteins are hardly detectable. In the case of the RdRp, larger amounts of the 3CD precursor and mature protein 3D are found in the infected cells, while only traces of the complete P3 block are detected. All of the 3C precursor molecules, such as 3CD, have an active protease that can catalyze the cleavages between the structural proteins [
45].
The most common naturally occurring cleavage sites of 3C proteases as P1-P1’ are Q-G, Q-S, and E-S, although Q-A, Q-C, or even Q-T and Q-I sequences are also cleaved in some cases. Both the different forms of the 3C proteases and the different sequences of the cleavage sites, appear to play regulatory roles. The same is true for the homologous 3C-like proteases of the other members of the order Picornavirales, which were named 3CL since the name 3C was reserved exclusively for the true picornaviruses (sensu stricto family Picornaviridae).
Although there are many exceptions, it can be stated that the 3CL proteases prefer (alpha-)glutaminyl residues at the P1 position (E and Q) and small neutral (G and S) or small hydrophobic amino acids (A) at the P1’ position [
46,
47]. A general sensitivity to thiol inhibitors such as NEM, Zn+, or iodoacetamide as well as a catalytic triad consisting of histidine, aspartate, and cysteine are, with minor exceptions, further common features among the 3CL proteases. Therefore, the 3C/3CL proteases are also classified together in the C3 peptidase family, which is therefore also called the picornain protease family, within clan PA (PROSITE entry PS51874,
https://prosite.expasy.org, accessed on 1 October 2023).
The processing of the nonstructural protein region and the responsible 3CL protease of members of the family
Iflaviridae have not been studied in much detail so far. However, several studies have investigated the enzymatic properties of the 3CL protease of various iflaviruses and have also begun to determine the boundaries of the mature 3CL protein. Ye and colleagues compared the 3CL sequence of the known iflaviruses [
26,
48,
49,
50,
51,
52] back in 2012 and identified a potential catalytic triad of H
2261, D
2299, and C
2383 in the Eov (amino acid numbers refer to the Ectropis obliqua virus, Genbank entry AY365064), which could be substantiated via mutational analyses in a bacterial expression system [
37]. However, the role of D
2299 could only be partially confirmed, as residual activity of the Eov 3CL-D
2299A was still observed. In the same study, the N- and C-termini of the 3CL protease of Eov were identified as A
2193 and Q
2491 using Edmann degradation revealing P1-P1’ cleavage site sequences of E-A and Q-S. A very comprehensive paper on DWV’s 3CL protease was published by Xuye Yuan and his colleague Tatsuhiko Kadowaki. They determined the N-terminus of DWV 3CL protease experimentally as G
2119 using a bacterial expression system and mutational analyses. Self-cleavage of a glutathione S-transferase (GST) 3CL fusion protein (GST-3CL) could be completely prevented if the cleavage site P1 residue Q
2118 was replaced with an alanine [
40]. The cleavage at the G
2119 residue and the corresponding Q-G cleavage site have been already predicted by Lanzi et al. in the initial description of the DWV genome [
26]. Furthermore, Yuan et al. used AlphaFold2 to model the structure of the 3CL protease of DWV and of many other insect- and iflaviruses, uncovering invariant cysteines and histidines as well as an unusual variety of different amino acids with an oxygen in the side chain as a third residue of the catalytic triads. Alanine substitution and enzyme studies provided additional evidence for an active center of the DWV 3CL protease containing the amino acids H
2170, C
2307, and N
2227, which were all pivotal for the enzymatic activity. A residual activity of 50% was observed if the conserved D
2225 was replaced by an alanine. Using affinity-purified rabbit antisera [
53], they detected the 3DL of DWV in infected bee pupal cells in Western blots as a 55 kDa protein and the 3CDL precursor as a 97 kDa protein. Furthermore, they generated a 3CL-specific rabbit antiserum, which showed a 42 kDa 3CL protease together with the 97 kDa 3CDL precursor. In another very recent publication, Palmer-Young and colleagues used a similar system to investigate the temperature sensitivity of DVW 3CL protease in relation to the honey bee’s life and honey bee’s temperature tolerance [
41].
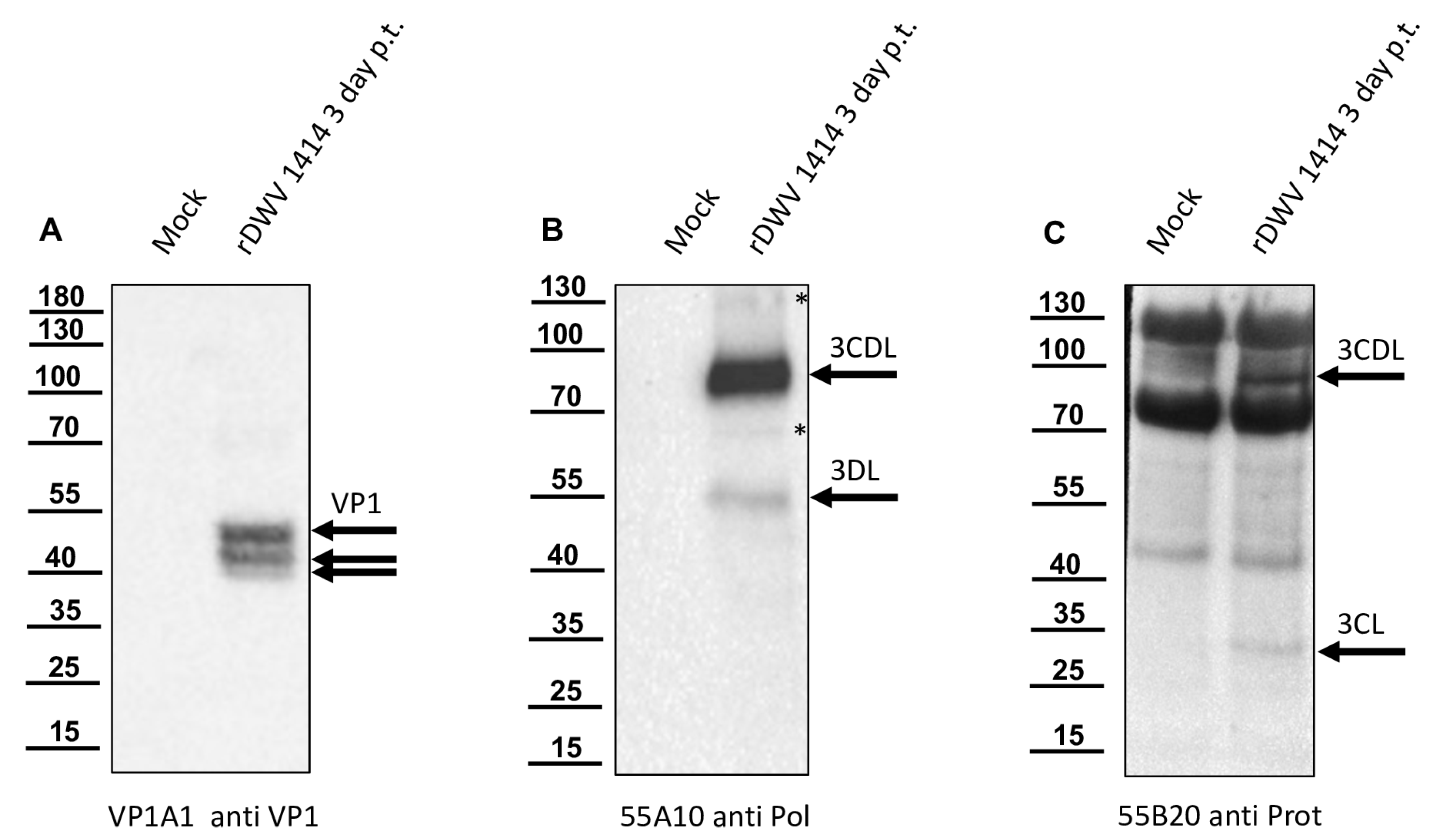
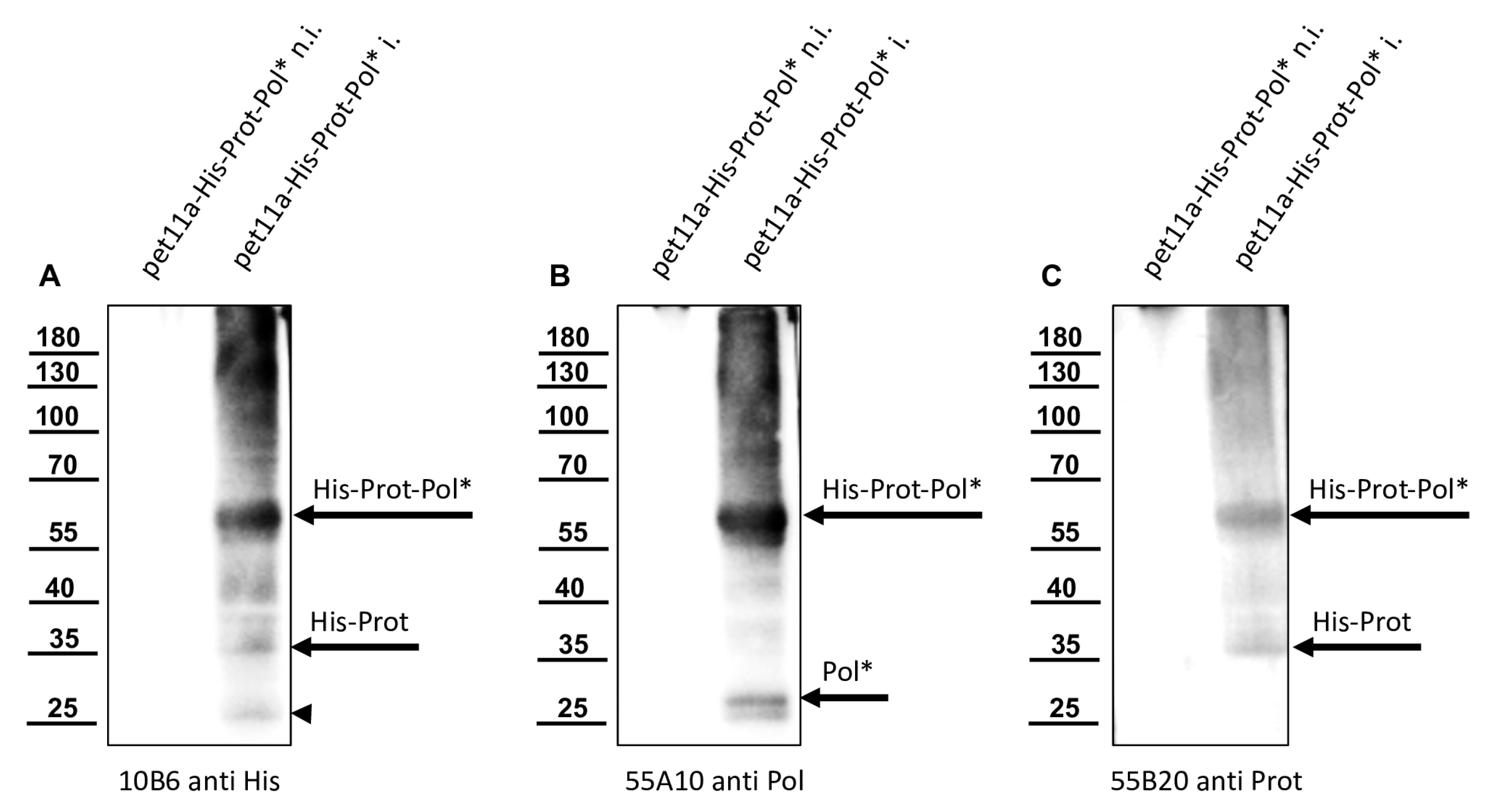
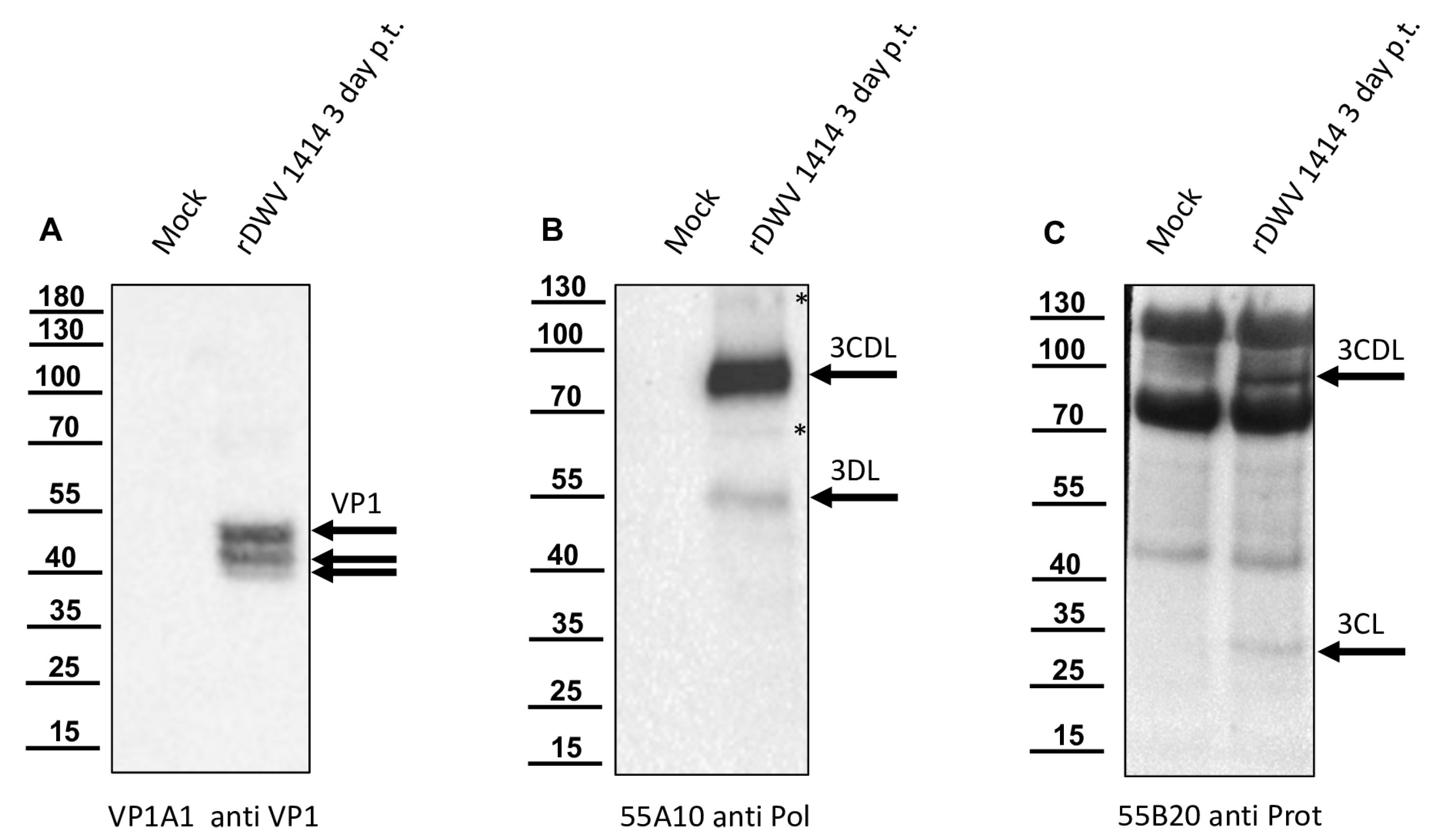
In this study, we aimed to elucidate the processing of the 3CL and 3DL region of DWV in more detail. We produced a 3CL-3DL polyprotein fragment in
E. coli and purified it via IMAC. This recombinant polyprotein fragment has been successfully used to generate mouse monoclonal antibodies that bind to epitopes in both 3CL and 3DL. Mab 55A10 anti-DWV detected a 3CDL precursor of 90 kDa as well as the mature 3DL with 55 kDa in Western blot analyses of DWV-infected bee pupae. Mab 55B20 anti-DWV detected the 3CDL precursor of 90 kDa and a mature DWV 3CL protease of about 32 kDa. The apparent molecular masses shown by our new monoclonal antibodies, obtained using a tricine-SDS-PAGE system, fit well with the experimentally determined boundaries of the proteins (see below). However, the pronounced differences between the molecular masses of these proteins with the proteins described by Yuan et al. are puzzling. Yuan et al. found a 3CDL precursor of 97 kDa and a mature 3CL protease with about 42 kDa, which appeared even larger when the size marker of
Figure 6C was examined in this publication [
40]. However, looking back to the original publication by Wu et al., in which the antiserum used against the RdRp of DWV was first presented, it can be seen that molecular weights of 90 (3CLDL) and 53 kDa (3DL) were reported there [
53].
We could show that the N-terminus of the DWV 3CL protease was self-cleaved between Q
2118 and G
2119 using Edmann degradation, just as predicted by Lanzi et al. on the basis of sequence comparisons [
26] and as confirmed by Yuan et al. using mutational analysis [
40]. We also identified the C-terminal residue of the mature 3CL protease by N-terminal sequencing of an RdRp fragment as Q
2393. To the best of our knowledge, the C-terminus of the DWV 3CL protease, and hence also the N-terminus of the DWV 3DL, have not been characterized before. Our refolded purified polyprotein fragment preparations were stable and not further self-processed during storage or incubation. This suggests that processing of the recombinant proteins occur mainly co-translationally in
E. coli, as Yuan et al. also observed for their GST-fusion protein [
40]. The N- and C-terminal self-cleavages of 3CL protease might represent inefficient, co-translational cis-events in the prokaryotic plasma but seem to be more efficient in the context of virus infection in the authentic polyprotein environment.
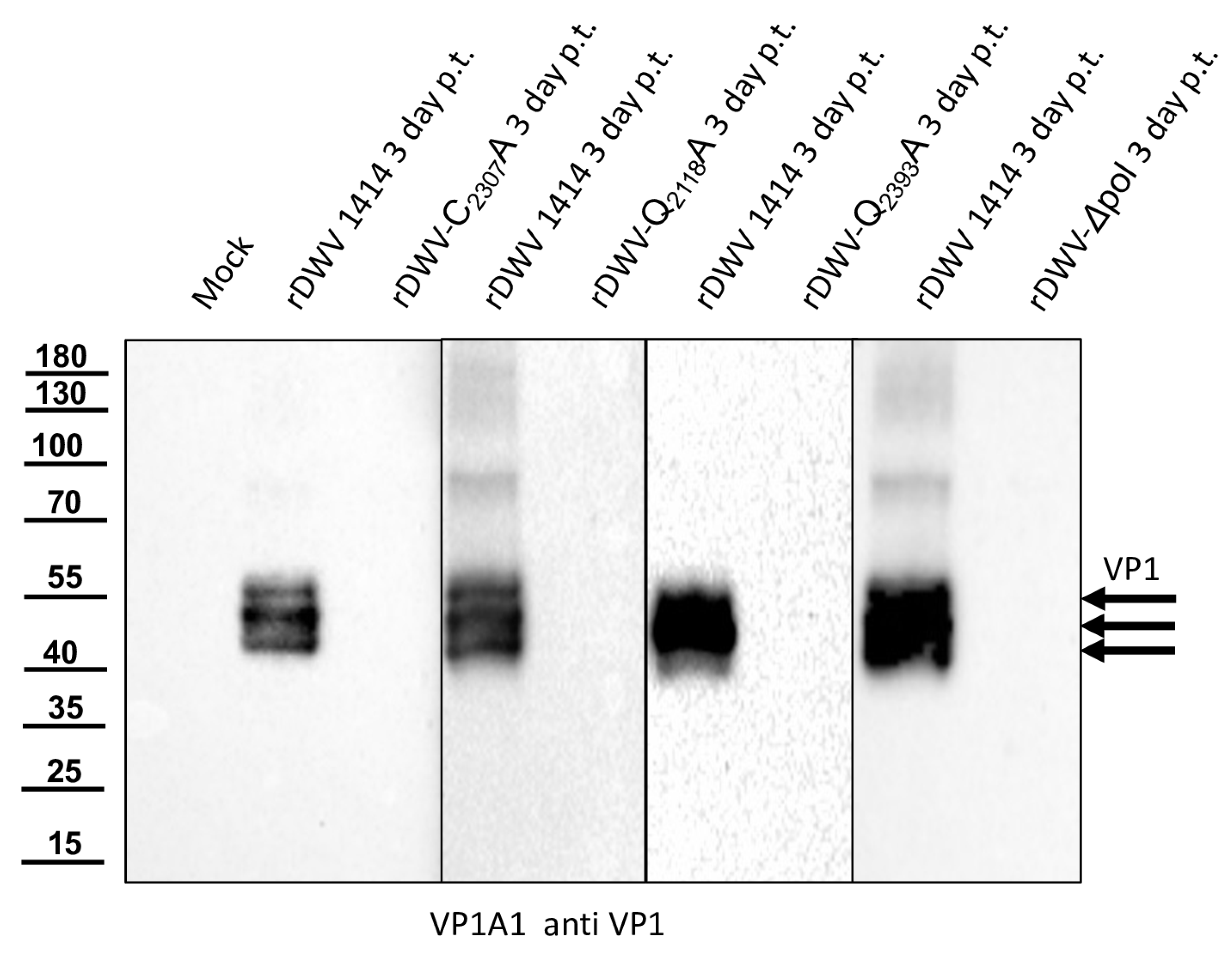
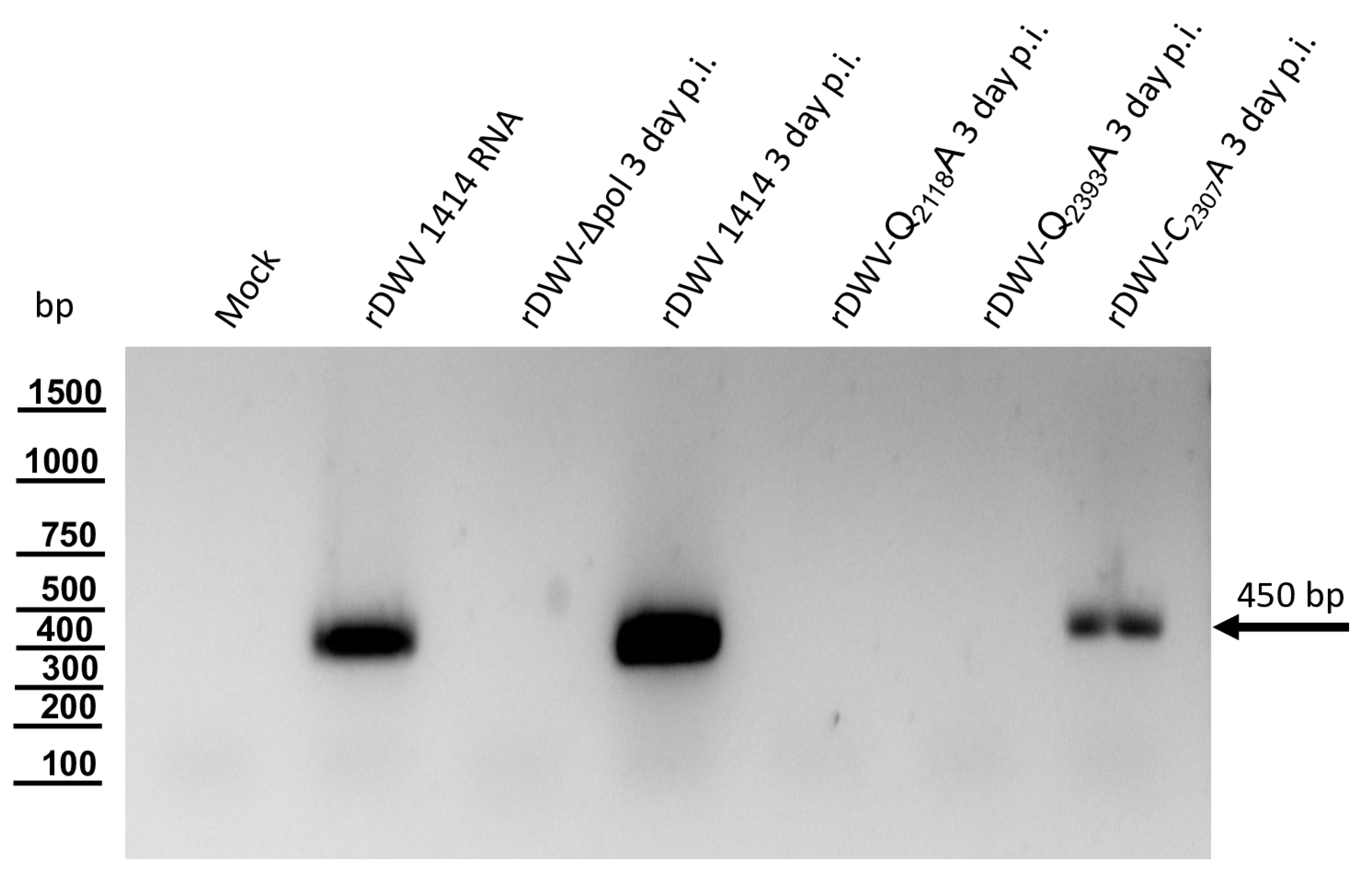
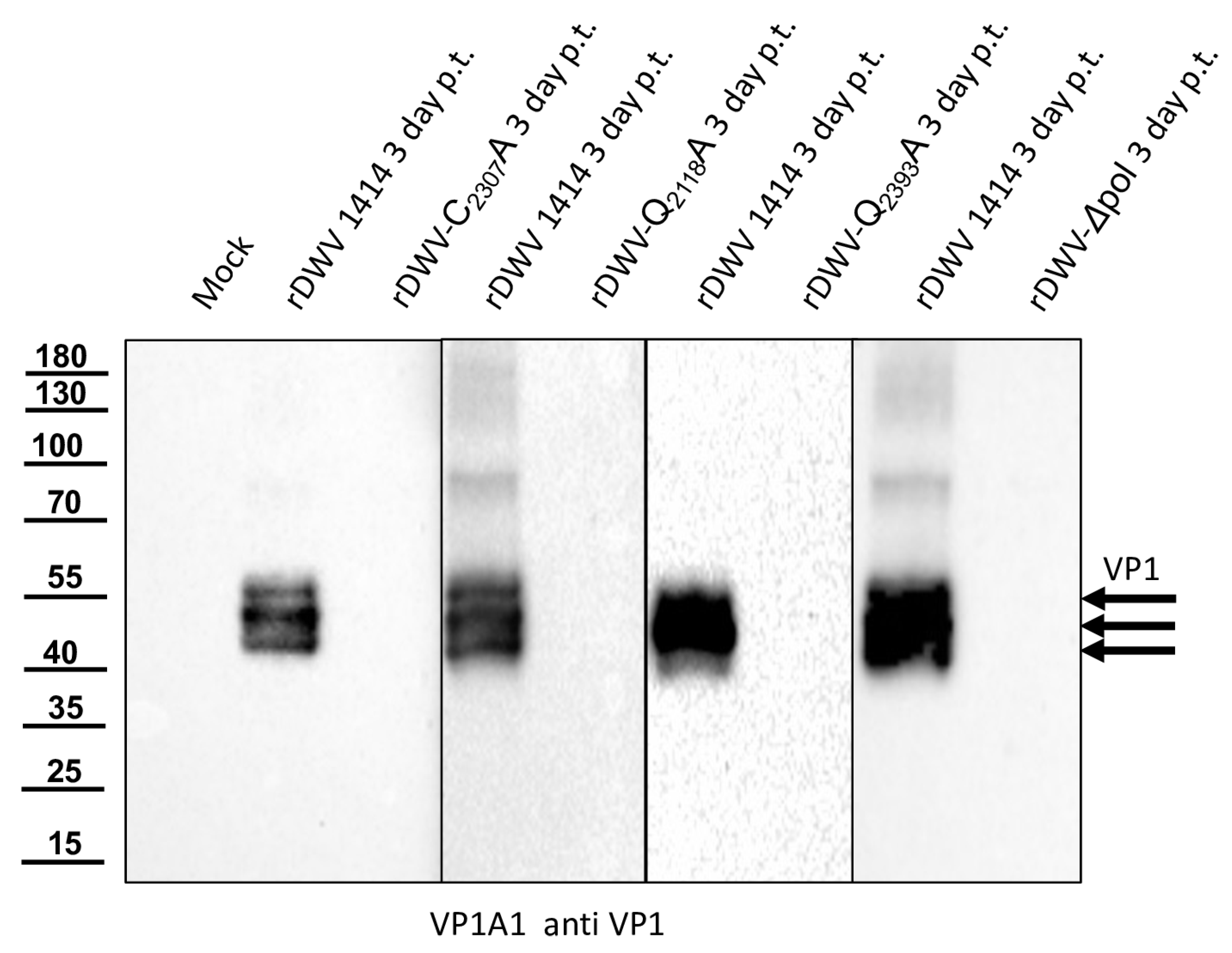
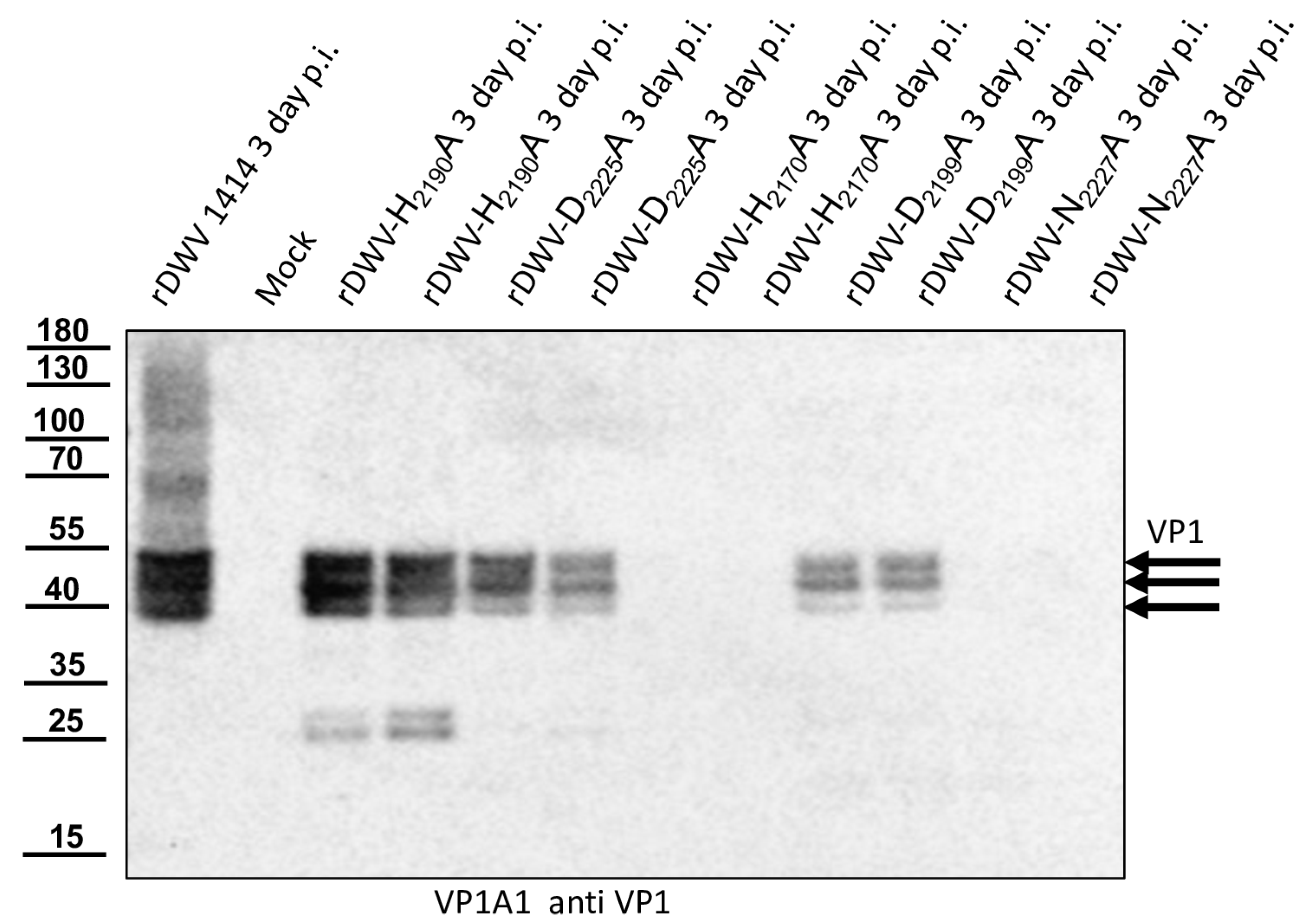
Our previous studies on molecular DWV clones have paved the way for additional analyses of the DWV 3CL protease in vivo. We verified our observations using reverse genetics and could demonstrate that 3CL protease activity and the identity of cleavage site residues are pivotal for DWV infection of honey bees. The exchange of the active cysteine (C
2307) and the exchanges of the two P1 glutamines (Q
2118 and Q
2393) against alanine prevented viral protein expression from being detected in the Western blot analysis after transfection of synthetic RNA. No replication or encapsidation of the synthetic input RNA was observed for both cleavage site mutants. Surprisingly, however, low-level replication and encapsidation of the synthetic RNA could be measured after transfection of the rDWV-C
2307A mutant. The 3CL protease domain of picornavirus-like agents adopts a chymotrypsin-like fold with a cysteine nucleophile in place of a commonly found serine. Serine-to-alanine exchange mutants of various serine protease molecules have already been shown to exhibit a residual activity. Evidence suggests that in these cases, a water molecule could serve as a poor substitute for the serine. Such a water molecule could be equipped with a nucleophilic attack character due to hydrogen bonding of the active histidine [
54,
55]. This could potentially explain why weak RNA replication still occurred in the rDWV-C
2307A mutant presented here. Whatever the case, the exchange of the active cysteine 2307 to an alanine resulted in a “non-viable” phenotype, because we could not detect VP1 expression and passage of the virus clone was not successful.
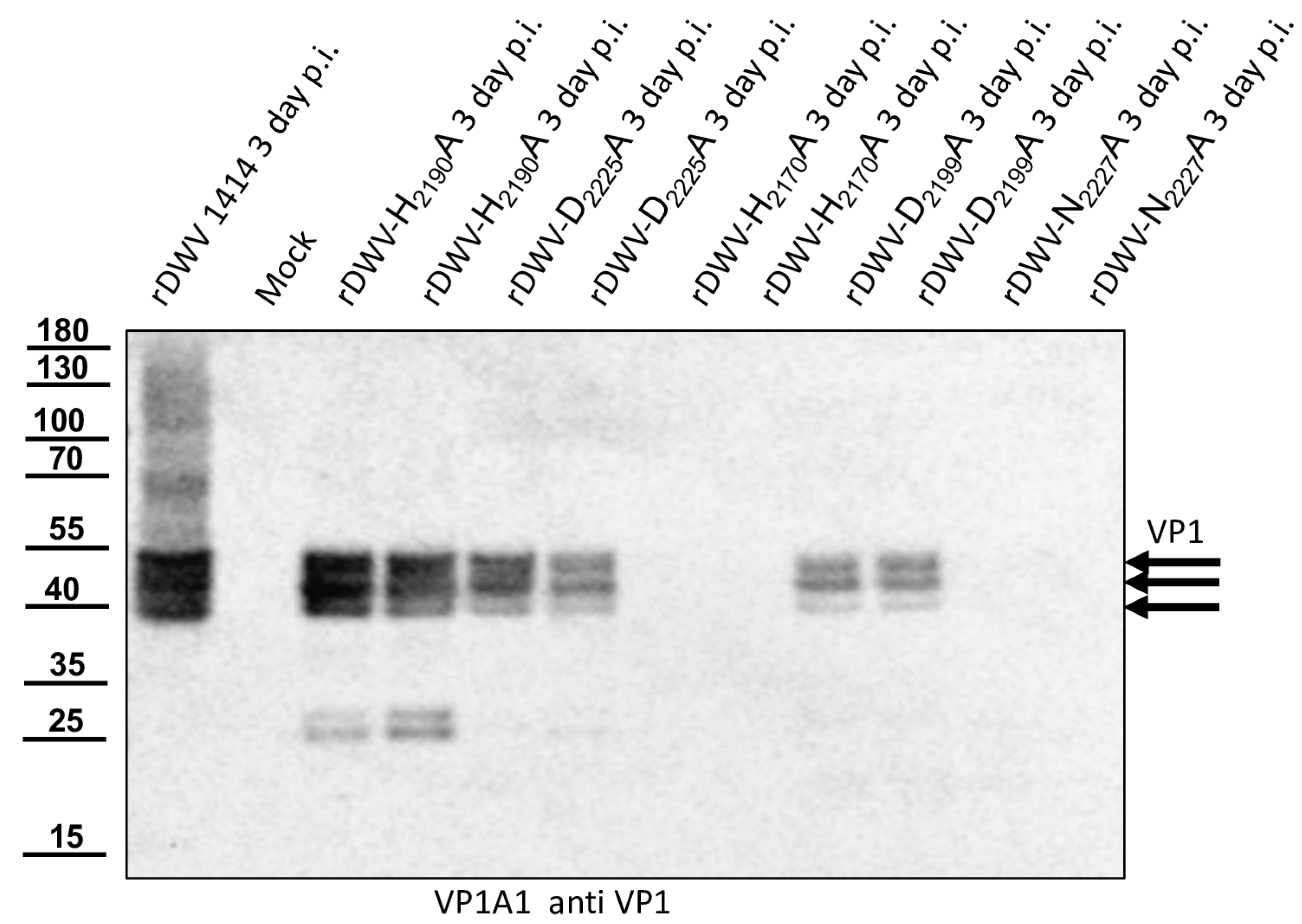
With this result, we decided to also investigate the other potential residues of the catalytic triad of the protease in vivo. Additional essential residues were found in H2170 and N2227 as postulated by Yuan et al. However, rDWV-H2190A and -D2225A showed an aberrant processing of VP1 and/or a significant reduction of viral protein expression, which was also observed in rDWV-D2199A. It must therefore be concluded that all these amino acid residues have a significance for the activity and/or specificity of the 3CL protease. The active-site residues of 3C proteases are in the cleft between the two barrels with a nucleophilic residue from the C-terminal barrel and a general acid basis with a histidine and glutamate or aspartate residues from the N-terminal barrel. The initial prediction of the catalytic triad of the DWV 3CL protease was carried out by Lanzi et al. solely based on sequence comparison with different ifla- and picornaviruses pointing at triad residues C2307, D2225, and H2190. Yuan et al. recently used the algorithms of AlphaFold2 to predict the protein structure of the 3CL protease of DWV and other insect viruses and found that the side chains of C2307, N2227, and H2170 form the active site of the 3C-like protease of DWV. Bacterial expression constructs with alanine substitutions of individual amino acids revealed that the identity of many amino acids of the 3C-like protease was pivotal for the activity in their cleavage assay. However, Yuan et al. were able to show that H2190 did not contribute to catalysis, as the exchange H2190A remained without functional effects in their assay. Amino acid D2225 was of minor importance, since an alanine exchange only reduced the 3CL activity by half. These analyses are conclusive, and asparagine fits well to a catalytic triad, which can be considered as being analogous to the Ser-His-Asp triad of known serine proteases. Our mutational analyses confirmed these hypotheses in vivo. We were able to inactivate the protease via targeted mutation of the suspected triad residues. However, mutation of other residues, such as D2225 and D2199, led to an obvious reduction in 3CL activity, restricted replication and reduced virus growth.
Future studies must now clarify to what extent the sequences of the cleavage sites, the protein structures and the cis- or trans-activity of 3CL protease influence the processing and replication of DWV in honey bees. In this study, we were at least able to clarify the boundaries of the 3CL protease beyond doubt, so that future studies can be carried out with the complete enzyme produced in the cell during infection.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}