
Genomic Markers Associated with Cytomegalovirus DNAemia in Kidney Transplant Recipients
 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Sequencing and Genotyping
2.3. Association and Meta-Association Analysis
2.4. Copy Number Variant Analysis
2.5. Extra Visualization and Downstream Analysis
3. Results
3.1. Study Population
3.2. Induction and Maintenance Immunosuppression Regimen
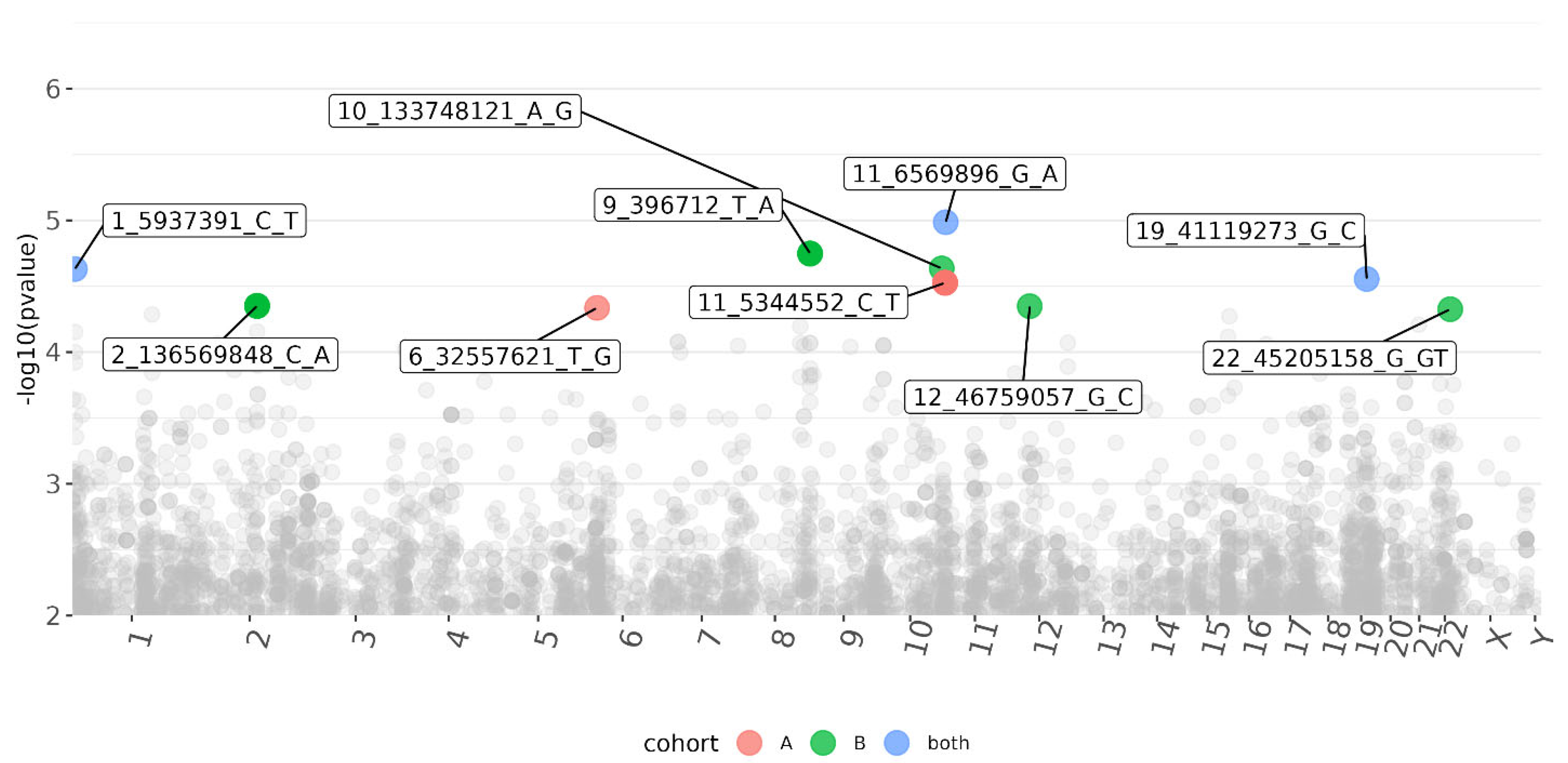
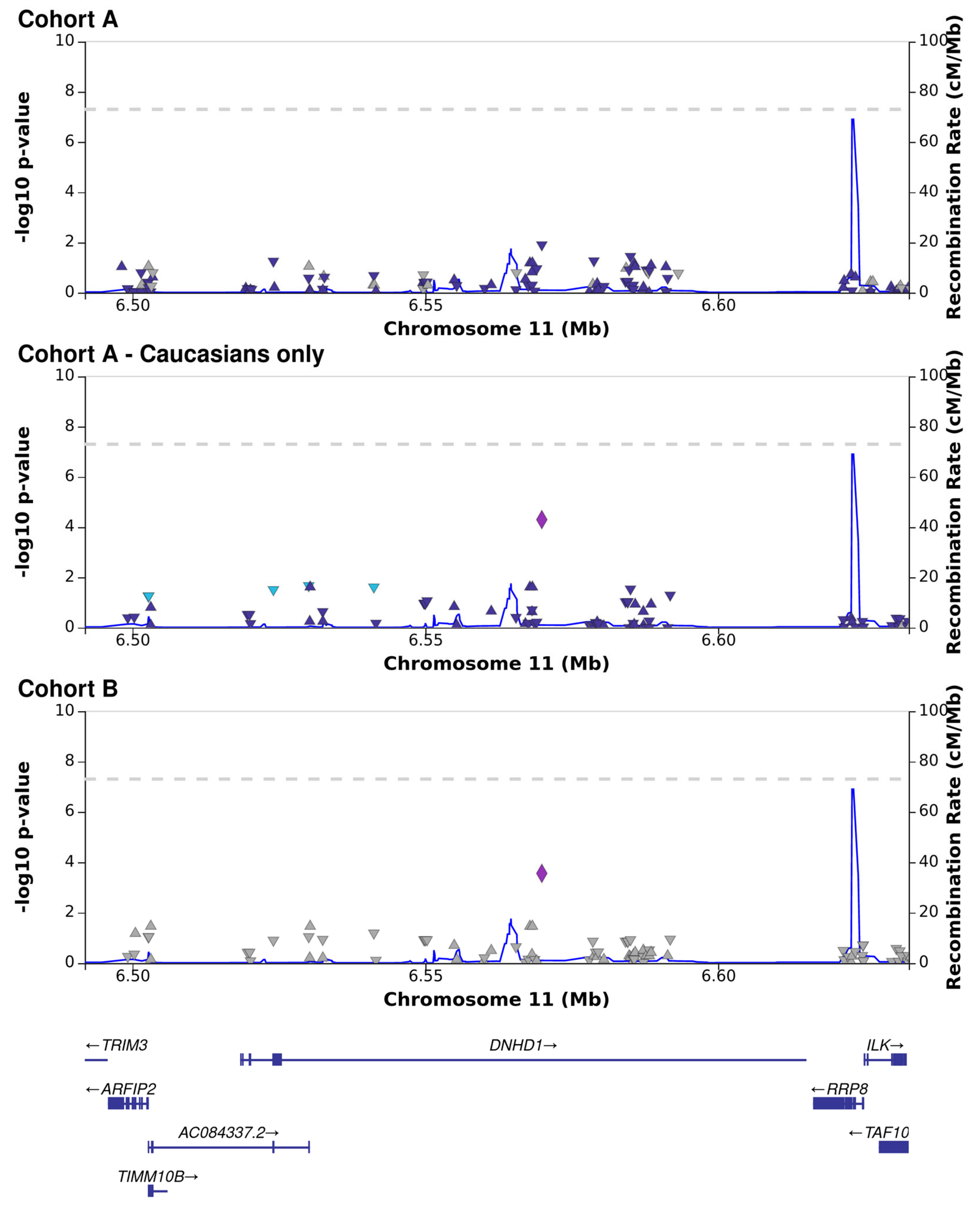
3.3. CMV DNAemia Variant Association Analysis
3.4. CMV DNAemia Copy Number Association Analysis
3.5. CMV DNAemia Peak Viral Load Association Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Staras, S.A.; Dollard, S.C.; Radford, K.W.; Flanders, W.D.; Pass, R.F.; Cannon, M.J. Seroprevalence of cytomegalovirus infection in the United States, 1988–1994. Clin. Infect. Dis. 2006, 43, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.D.; Clark, D.A.; Emery, V.C. Betaherpesviruses in transplant recipients. J. Antimicrob. Chemother. 2000, 45 (Suppl. T3), 29–34. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, A.; Schluchter, M.; Easley, K.; Demmler, G.; Shearer, W.; La, R.P.; Pitt, J.; Cooper, E.; Goldfarb, J.; Hodes, D.; et al. Cytomegalovirus infection and HIV-1 disease progression in infants born to HIV-1-infected women. Pediatric Pulmonary and Cardiovascular Complications of Vertically Transmitted HIV Infection Study Group. N. Engl. J. Med. 1999, 341, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R.; Humar, A. Cytomegalovirus in solid organ transplant recipients-Guidelines of the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transpl. 2019, 33, e13512. [Google Scholar] [CrossRef] [PubMed]
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Huprikar, S.; Chou, S.; Danziger-Isakov, L.; Humar, A.; The Transplantation Society International CMV Consensus Group. The Third International Consensus Guidelines on the Management of Cytomegalovirus in Solid-organ Transplantation. Transplantation 2018, 102, 900–931. [Google Scholar] [CrossRef] [PubMed]
- De Keyzer, K.; Van Laecke, S.; Peeters, P.; Vanholder, R. Human cytomegalovirus and kidney transplantation: A clinician’s update. Am. J. Kidney Dis. 2011, 58, 118–126. [Google Scholar] [CrossRef] [PubMed]
- De Matos, S.B.; Meyer, R.; Lima, F.W.M. Cytomegalovirus Infection after Renal Transplantation: Occurrence, Clinical Features, and the Cutoff for Antigenemia in a University Hospital in Brazil. Infect. Chemother. 2017, 49, 255–261. [Google Scholar] [CrossRef]
- Hodson, E.M.; Ladhani, M.; Webster, A.C.; Strippoli, G.F.; Craig, J.C. Antiviral medications for preventing cytomegalovirus disease in solid organ transplant recipients. Cochrane Database Syst. Rev. 2013, 2, CD003774. [Google Scholar] [CrossRef] [PubMed]
- Arthurs, S.K.; Eid, A.J.; Pedersen, R.A.; Kremers, W.K.; Cosio, F.G.; Patel, R.; Razonable, R.R. Delayed-onset primary cytomegalovirus disease and the risk of allograft failure and mortality after kidney transplantation. Clin. Infect. Dis. 2008, 46, 840–846. [Google Scholar] [CrossRef]
- Vink, C.; Smit, M.J.; Leurs, R.; Bruggeman, C.A. The role of cytomegalovirus-encoded homologs of G protein-coupled receptors and chemokines in manipulation of and evasion from the immune system. J. Clin. Virol. 2001, 23, 43–55. [Google Scholar] [CrossRef]
- Wang, D.; Bresnahan, W.; Shenk, T. Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 16642–16647. [Google Scholar] [CrossRef] [PubMed]
- Penfold, M.E.; Dairaghi, D.J.; Duke, G.M.; Saederup, N.; Mocarski, E.S.; Kemble, G.W.; Schall, T.J. Cytomegalovirus encodes a potent alpha chemokine. Proc. Natl. Acad. Sci. USA 1999, 96, 9839–9844. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 1996, 70, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Kijpittayarit, S.; Eid, A.J.; Brown, R.A.; Paya, C.V.; Razonable, R.R. Relationship between Toll-like receptor 2 polymorphism and cytomegalovirus disease after liver transplantation. Clin. Infect. Dis. 2007, 44, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, P.; Schaeffeler, E.; Witzke, O.; Nitschke, M.; Kliem, V.; Zortel, M.; Wagner, E.-M.; Schwab, M.; Hauser, I.A. No association of genetic variants in TLR4, TNF-alpha, IL10, IFN-gamma, and IL37 in cytomegalovirus-positive renal allograft recipients with active CMV infection-Subanalysis of the prospective randomised VIPP study. PLoS ONE 2021, 16, e0246118. [Google Scholar] [CrossRef] [PubMed]
- Futohi, F.; Saber, A.; Nemati, E.; Einollahi, B.; Rostami, Z. Human Leukocyte Antigen Alleles and Cytomegalovirus Infection After Renal Transplantation. Nephrourol. Mon. 2015, 7, e31635. [Google Scholar] [CrossRef] [PubMed]
- Guberina, H.; da Silva Nardi, F.; Michita, R.T.; Dolff, S.; Bienholz, A.; Heinemann, F.M.; Wilde, B.; Trilling, M.; Horn, P.A.; Kribben, A.; et al. Susceptibility of HLA-E*01:03 Allele Carriers to Develop Cytomegalovirus Replication After Living-Donor Kidney Transplantation. J. Infect. Dis. 2018, 217, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Pos, O.; Radvanszky, J.; Buglyo, G.; Pos, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef]
- Gonzalez, E.; Kulkarni, H.; Bolivar, H.; Mangano, A.; Sanchez, R.; Catano, G.; Nibbs, R.J.; Freedman, B.I.; Quinones, M.P.; Bamshad, M.J.; et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 2005, 307, 1434–1440. [Google Scholar] [CrossRef]
- Cao, K.; Marin, D.; Sekine, T.; Rondon, G.; Zhao, W.; Smith, N.T.; Daher, M.; Wang, Q.; Li, L.; Saliba, R.M.; et al. Donor NKG2C Copy Number: An Independent Predictor for CMV Reactivation After Double Cord Blood Transplantation. Front. Immunol. 2018, 9, 2444. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Sham, P.C.; Purcell, S.M. Statistical power and significance testing in large-scale genetic studies. Nat. Rev. Genet. 2014, 15, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Boughton, A.P.; Welch, R.P.; Flickinger, M.; VandeHaar, P.; Taliun, D.; Abecasis, G.R.; Boehnke, M. LocusZoom.js: Interactive and embeddable visualization of genetic association study results. Bioinformatics 2021, 37, 3017–3018. [Google Scholar] [CrossRef] [PubMed]
- Fadista, J.; Manning, A.K.; Florez, J.C.; Groop, L. The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur. J. Hum. Genet. 2016, 24, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R.; Humar, A. Cytomegalovirus in solid organ transplantation. Am. J. Transplant. 2013, 13 (Suppl. 4), 93–106. [Google Scholar] [CrossRef] [PubMed]
- Merino-Gracia, J.; Garcia-Mayoral, M.F.; Rodriguez-Crespo, I. The association of viral proteins with host cell dynein components during virus infection. FEBS J. 2011, 278, 2997–3011. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Yu, Y.; Pierciey, F.J., Jr.; Alwine, J.C. Human cytomegalovirus induces the endoplasmic reticulum chaperone BiP through increased transcription and activation of translation by using the BiP internal ribosome entry site 11. J. Virol. 2010, 84, 11479–11486. [Google Scholar] [CrossRef]
- Clippinger, A.J.; Alwine, J.C. Dynein mediates the localization and activation of mTOR in normal and human cytomegalovirus-infected cells. Genes Dev. 2012, 26, 2015–2026. [Google Scholar] [CrossRef]
- Alwine, J.C. The human cytomegalovirus assembly compartment: A masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog. 2012, 8, e1002878. [Google Scholar] [CrossRef]
- Dohner, K.; Wolfstein, A.; Prank, U.; Echeverri, C.; Dujardin, D.; Vallee, R.; Sodeik, B. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 2002, 13, 2795–2809. [Google Scholar] [CrossRef] [PubMed]
- Hancock, M.H.; Crawford, L.B.; Pham, A.H.; Mitchell, J.; Struthers, H.M.; Yurochko, A.D.; Caposio, P.; Nelson, J.A. Human Cytomegalovirus miRNAs Regulate TGF-beta to Mediate Myelosuppression while Maintaining Viral Latency in CD34(+) Hematopoietic Progenitor Cells. Cell Host Microbe 2020, 27, 104–114.e4. [Google Scholar] [CrossRef] [PubMed]
- Alcami, J.; Paya, C.V.; Virelizier, J.L.; Michelson, S. Antagonistic modulation of human cytomegalovirus replication by transforming growth factor beta and basic fibroblastic growth factor. J. Gen. Virol. 1993, 74 Pt 2, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Michelson, S.; Alcami, J.; Kim, S.J.; Danielpour, D.; Bachelerie, F.; Picard, L.; Bessia, C.; Paya, C.; Virelizier, J.L. Human cytomegalovirus infection induces transcription and secretion of transforming growth factor beta 1. J. Virol. 1994, 68, 5730–5737. [Google Scholar] [CrossRef] [PubMed]
- Helantera, I.; Teppo, A.M.; Koskinen, P.; Tornroth, T.; Gronhagen-Riska, C.; Lautenschlager, I. Increased urinary excretion of transforming growth factor-beta(1) in renal transplant recipients during cytomegalovirus infection. Transpl. Immunol. 2006, 15, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Helantera, I.; Loginov, R.; Koskinen, P.; Tornroth, T.; Gronhagen-Riska, C.; Lautenschlager, I. Persistent cytomegalovirus infection is associated with increased expression of TGF-beta1, PDGF-AA and ICAM-1 and arterial intimal thickening in kidney allografts. Nephrol. Dial. Transpl. 2005, 20, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, M.; Murphy-Ullrich, J.E.; Britt, W.J. Human cytomegalovirus induces TGF-beta1 activation in renal tubular epithelial cells after epithelial-to-mesenchymal transition. PLoS Pathog. 2010, 6, e1001170. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Liu, J.; Gu, J.; Zhu, P. HLA-DRB1*09 is associated with increased incidence of cytomegalovirus infection and disease after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transpl. 2007, 13, 1417–1421. [Google Scholar] [CrossRef]
- Varga, M.; Rajczy, K.; Telkes, G.; Hidvegi, M.; Peter, A.; Remport, A.; Korbonits, M.; Fazakas, J.; Toronyi, É.; Sárváry, E.; et al. HLA-DQ3 is a probable risk factor for CMV infection in high-risk kidney transplant patients. Nephrol. Dial. Transpl. 2008, 23, 2673–2678. [Google Scholar] [CrossRef]
- Rice, A.M.; McLysaght, A. Dosage sensitivity is a major determinant of human copy number variant pathogenicity. Nat. Commun. 2017, 8, 14366. [Google Scholar] [CrossRef] [PubMed]
- Ionita-Laza, I.; Rogers, A.J.; Lange, C.; Raby, B.A.; Lee, C. Genetic association analysis of copy-number variation (CNV) in human disease pathogenesis. Genomics 2009, 93, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Dai, X.; Xiao, Y.; Du, Y.; Chapa, T.J.; Johnson, J.R.; Li, X.; Krogan, N.J.; Deng, H.; Wu, T.-T.; et al. Virus-Like Vesicles of Kaposi’s Sarcoma-Associated Herpesvirus Activate Lytic Replication by Triggering Differentiation Signaling. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Duan, R.; Gao, Y.; Zhang, H.; Qiao, Y.; Li, Q.; Zhao, F. Role of Epstein-Barr Virus and Human Papillomavirus Coinfection in Cervical Intraepithelial Neoplasia in Chinese Women Living With HIV. Front. Cell. Infect. Microbiol. 2021, 11, 703259. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Matsuda, K.; Tanikawa, C.; Lin, J.; Furukawa, Y.; Hamamoto, R.; Nakamura, Y. Late Cornified Envelope Group I, a novel target of p53, regulates PRMT5 activity. Neoplasia 2014, 16, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Kuenzel, S.; Till, A.; Winkler, M.; Hasler, R.; Lipinski, S.; Jung, S.; Grötzinger, J.; Fickenscher, H.; Schreiber, S.; Rosenstiel, P. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J. Immunol. 2010, 184, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Tangden, T.; Gustafsson, S.; Rao, A.S.; Ingelsson, E. A genome-wide association study in a large community-based cohort identifies multiple loci associated with susceptibility to bacterial and viral infections. Sci. Rep. 2022, 12, 2582. [Google Scholar] [CrossRef] [PubMed]
- Butković, A.; Elena, S.F. Genome-wide association studies of viral infections—A short guide to a successful experimental and statistical analysis. Front. Syst. Biol. 2022, 2, 1005758. [Google Scholar] [CrossRef]
- Casto, A.M.; Seo, S.; Levine, D.M.; Storer, B.E.; Dong, X.; Hansen, J.A.; Boeckh, M.; Martin, P.J. Genetic variants associated with cytomegalovirus infection after allogeneic hematopoietic cell transplantation. Blood 2021, 138, 1628–1636. [Google Scholar] [CrossRef]
- Duchowny, K.A.; Noppert, G.A. The Association Between Cytomegalovirus and Disability by Race/Ethnicity and Sex: Results From the Health and Retirement Study. Am. J. Epidemiol. 2021, 190, 2314–2322. [Google Scholar] [CrossRef]
- Su, F.; Green, P.K.; Berry, K.; Ioannou, G.N. The association between race/ethnicity and the effectiveness of direct antiviral agents for hepatitis C virus infection. Hepatology 2017, 65, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Randolph, H.E.; Fiege, J.K.; Thielen, B.K.; Mickelson, C.K.; Shiratori, M.; Barroso-Batista, J.; Langlois, R.A.; Barreiro, L.B. Genetic ancestry effects on the response to viral infection are pervasive but cell type specific. Science 2021, 374, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Kist, N.C.; Lambert, B.; Campbell, S.; Katzourakis, A.; Lunn, D.; Lemey, P.; Iversen, A.K.N. HIV-1 p24Gag adaptation to modern and archaic HLA-allele frequency differences in ethnic groups contributes to viral subtype diversification. Virus Evol. 2020, 6, veaa085. [Google Scholar] [CrossRef]
- Shapira, G.; Shomron, N.; Gurwitz, D. Ethnic differences in alpha-1 antitrypsin deficiency allele frequencies may partially explain national differences in COVID-19 fatality rates. FASEB J. 2020, 34, 14160–14165. [Google Scholar] [CrossRef] [PubMed]
- Banday, A.R.; Stanifer, M.L.; Florez-Vargas, O.; Onabajo, O.O.; Papenberg, B.W.; Zahoor, M.A.; Mirabello, L.; Ring, T.J.; Lee, C.-H.; Albert, P.S.; et al. Genetic regulation of OAS1 nonsense-mediated decay underlies association with COVID-19 hospitalization in patients of European and African ancestries. Nat. Genet. 2022, 54, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Namkoong, H.; Edahiro, R.; Takano, T.; Nishihara, H.; Shirai, Y.; Sonehara, K.; Tanaka, H.; Azekawa, S.; Mikami, Y.; Lee, H.; et al. DOCK2 is involved in the host genetics and biology of severe COVID-19. Nature 2022, 609, 754–760. [Google Scholar] [CrossRef]
- Gurdasani, D.; Barroso, I.; Zeggini, E.; Sandhu, M.S. Genomics of disease risk in globally diverse populations. Nat. Rev. Genet. 2019, 20, 520–535. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, H.; Yuan, X.; Gao, K.; Duan, J. Comparative study of whole exome sequencing-based copy number variation detection tools. BMC Bioinform. 2020, 21, 97. [Google Scholar] [CrossRef]
- Fisher, S.; Barry, A.; Abreu, J.; Minie, B.; Nolan, J.; Delorey, T.M.; Young, G.; Fennell, T.J.; Allen, A.; Ambrogio, L.; et al. A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 2011, 12, R1. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v1. [Google Scholar]
- Church, D.M.; Schneider, V.A.; Graves, T.; Auger, K.; Cunningham, F.; Bouk, N.; Chen, H.C.; Agarwala, R.; McLaren, W.M.; Ritchie, G.R.; et al. Modernizing reference genome assemblies. PLoS Biol. 2011, 9, e1001091. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 43, 931–945. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Exome Aggregation Consortium (ExAC), Cambridge, MA, USA. Available online: http://exac.broadinstitute.org (accessed on 4 October 2023).
- 1000 Genomes Project Consortium; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; McVean, G.A. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [PubMed]
- Fritz, M.H.Y.; Leinonen, R.; Cochrane, G.; Birney, E. Efficient storage of high throughput DNA sequencing data using reference-based compression. Genome Res. 2011, 21, 734–740. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Cohort | A | B | Total |
|---|---|---|---|
| Participants (%) | 72 (36.3%) | 126 (63.6%) | 198 (100%) |
| Male (%) | 41 (56.9%) | 75 (59.5%) | 116 (58.5%) |
| CMV DNAemia (%) | 36 (50%) | 60 (47.6%) | 96 (48.4%) |
| Caucasian (%) | 54 (75%) | 126 (100%) | 180 (90.9%) |
| Age at time of transplant (Mean, SE) | 54.5 (14) | 51.2 (1.4) | 51.2 (1.4) |
| Peak viral load (Mean, SE) | 2.4 × 106 (1.4 × 106) | 6.5 × 105 (2.6 × 105) | 1.3 × 106 (5.5 × 106) |
| Variant | p-Value | Beta | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | GENE | EFFECT | CADD | Meta | A | B | Z-Meta | A | B |
| 11_6569896_G_A | DNHD1 | Frameshift, missense | 23.5 | 1.00 × 10−5 | 1.20 × 10−2 | 4.90 × 10−4 | −4.41 | −0.25 | −0.28 |
| 1_5937391_C_T | NPHP4 | Intron | 0.184 | 2.30 × 10−5 | 5.40 × 10−3 | 1.10 × 10−3 | 4.23 | 0.3 | 0.24 |
| 19_41119273_G_C | LTBP4 | Intron | 6.661 | 2.80 × 10−5 | 3.20 × 10−3 | 1.70 × 10−3 | −4.19 | −0.26 | −0.24 |
| 22_45205158_G_GT | PRR5—ARHGAP8 | Intron | 2.397 | 2.80 × 10−5 | N/A | 4.70 × 10−5 | 4.19 | N/A | 0.38 |
| 6_32557621_T_G | HLA-DRB1 | Upstream | N/A | 4.60 × 10−5 | 4.60 × 10−5 | N/A | 4.07 | 0.51 | N/A |
| 1_166591271_T_C | FMO9P | Splice donor and intron | 23.2 | 5.20 × 10−5 | 9.60 × 10−3 | 1.30 × 10−3 | 4.05 | 0.34 | 0.25 |
| 21_27840567_C_T | CYYR1 | 3′ prime UTR | 7.693 | 6.20 × 10−5 | 6.20 × 10−5 | N/A | −4.01 | −0.58 | NA |
| 8_126085586_G_A | WASHC5 | Intron | 1.184 | 6.40 × 10−5 | 5.90 × 10−3 | 2.70 × 10−3 | −4 | −0.42 | −0.25 |
| 8_126068873_T_G | WASHC5 | Intron | 0.375 | 8.50 × 10−5 | 2.40 × 10−2 | 1.10 × 10−3 | −3.93 | −0.43 | −0.36 |
| 10_12195881_G_A | SEC61A2 | Intron | 3.243 | 8.90 × 10−5 | 8.90 × 10−5 | N/A | −3.92 | −0.37 | N/A |
| SYMBOL | A_Loss | B_Loss | A_Gain | B_Gain | A | B | Loss | Gain | Overall | FDR |
|---|---|---|---|---|---|---|---|---|---|---|
| LCE3B | 12 | 16 | 0 | 0 | 12 | 16 | 28 | 0 | 28 | 2.51 × 10−6 |
| LCE3C | 13 | 15 | 0 | 0 | 13 | 15 | 28 | 0 | 28 | 2.51 × 10−6 |
| TAS2R43 | 5 | 13 | 0 | 0 | 5 | 13 | 18 | 0 | 18 | 1.71 × 10−3 |
| GSTM1 | 17 | 0 | 0 | 0 | 17 | 0 | 17 | 0 | 17 | 2.06 × 10−3 |
| GSTM2 | 17 | 0 | 0 | 0 | 17 | 0 | 17 | 0 | 17 | 2.06 × 10−3 |
| AHNAK2 | 9 | 3 | 0 | 0 | 9 | 3 | 12 | 0 | 12 | 2.99 × 10−2 |
| ID | GENE | EFFECT | p-Value | Beta | CADD |
|---|---|---|---|---|---|
| 13_23808782_T_C | SGCG | Frameshift and missense | 1.10 × 10−5 | 5.21 | 5.754 |
| 11_133788869_A_G | IGSF9B | Intron | 1.80 × 10−5 | −3.01 | 0.173 |
| 17_39394962_C_T | KRTAP9-8 | 3_prime_UTR|intron | 2.00 × 10−5 | 2.86 | 3.321 |
| 2_112939548_T_C | FBLN7 | Intron | 2.10 × 10−5 | 5.07 | 6.004 |
| 2_112940578_C_T | FBLN7 | Intron | 2.10 × 10−5 | 5.07 | 1.94 |
| 6_36292007_G_A | BNIP5 | Intron | 2.10 × 10−5 | 4.14 | 0.052 |
| 16_57068107_C_T | NLRC5 | Frameshift and missense | 2.60 × 10−5 | −4.42 | 1.901 |
| 16_57071209_T_C | NLRC5 | Intron | 3.40 × 10−5 | −3.92 | 1.492 |
| 16_57071226_TCC_T | NLRC5 | Intron | 3.40 × 10−5 | −3.92 | 4.507 |
| 16_57071236_C_T | NLRC5 | Intron | 3.40 × 10−5 | −3.92 | 4.909 |
| 15_59499179_G_A | LDHAL6B|MYO1E | Frameshift and missense | 3.50 × 10−5 | 2.86 | 8.694 |
| 15_59500116_T_C | LDHAL6B|MYO1E | frameshift and missense | 3.50 × 10−5 | 2.86 | 23.1 |
| 16_57075406_T_C | NLRC5 | Frameshift and missense | 3.60 × 10−5 | −4.48 | 8.512 |
| 16_57076018_G_C | NLRC5 | Intron | 3.60 × 10−5 | −4.48 | 0.213 |
| 16_57077523_G_A | NLRC5 | Splice region and intron | 3.60 × 10−5 | −4.48 | 2.641 |
| 16_57077581_C_T | NLRC5 | Intron | 3.60 × 10−5 | −4.48 | 0.278 |
| 16_57060213_C_T | NLRC5 | Frameshift and missense | 3.60 × 10−5 | −4.48 | 3.843 |
| 16_57060340_T_C | NLRC5 | Frameshift and missense | 3.60 × 10−5 | −4.48 | 5.063 |
| 16_57060353_T_C | NLRC5 | Frameshift and missense | 3.60 × 10−5 | −4.48 | 0.581 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shapira, G.; Volkov, H.; Fabian, I.; Mohr, D.W.; Bettinotti, M.; Shomron, N.; Avery, R.K.; Arav-Boger, R. Genomic Markers Associated with Cytomegalovirus DNAemia in Kidney Transplant Recipients. Viruses 2023, 15, 2227. https://doi.org/10.3390/v15112227
Shapira G, Volkov H, Fabian I, Mohr DW, Bettinotti M, Shomron N, Avery RK, Arav-Boger R. Genomic Markers Associated with Cytomegalovirus DNAemia in Kidney Transplant Recipients. Viruses. 2023; 15(11):2227. https://doi.org/10.3390/v15112227
Chicago/Turabian StyleShapira, Guy, Hadas Volkov, Itai Fabian, David W. Mohr, Maria Bettinotti, Noam Shomron, Robin K. Avery, and Ravit Arav-Boger. 2023. "Genomic Markers Associated with Cytomegalovirus DNAemia in Kidney Transplant Recipients" Viruses 15, no. 11: 2227. https://doi.org/10.3390/v15112227
APA StyleShapira, G., Volkov, H., Fabian, I., Mohr, D. W., Bettinotti, M., Shomron, N., Avery, R. K., & Arav-Boger, R. (2023). Genomic Markers Associated with Cytomegalovirus DNAemia in Kidney Transplant Recipients. Viruses, 15(11), 2227. https://doi.org/10.3390/v15112227