
Genomic and Epidemiologic Surveillance of SARS-CoV-2 in the Pandemic Period: Sequencing Network of the Lazio Region, Italy

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling Strategy
- I.
- Monthly flash surveys: the Istituto Superiore di Sanità (ISS) communicated the monthly collection day and the relative sample size, SeRESMI calculated the geographical distribution within the Lazio region and the INMI laboratory indicated to the diagnostic laboratories network (CoroNET-Lazio) the modalities and the number of samples requiring sequencing by one of the WGSnet laboratories;
- II.
- Continuous flow of weekly sequencing: in this case, the sampling, instead of being random, was targeted at hospitalized patients with severe COVID-19 disease and/or persistent SARS-CoV-2 infection and the total number of samples to be sequenced was based on ISS indications.
2.2. Sequencing
2.3. Sequence Analysis
2.4. Epidemiological Analysis
3. Results
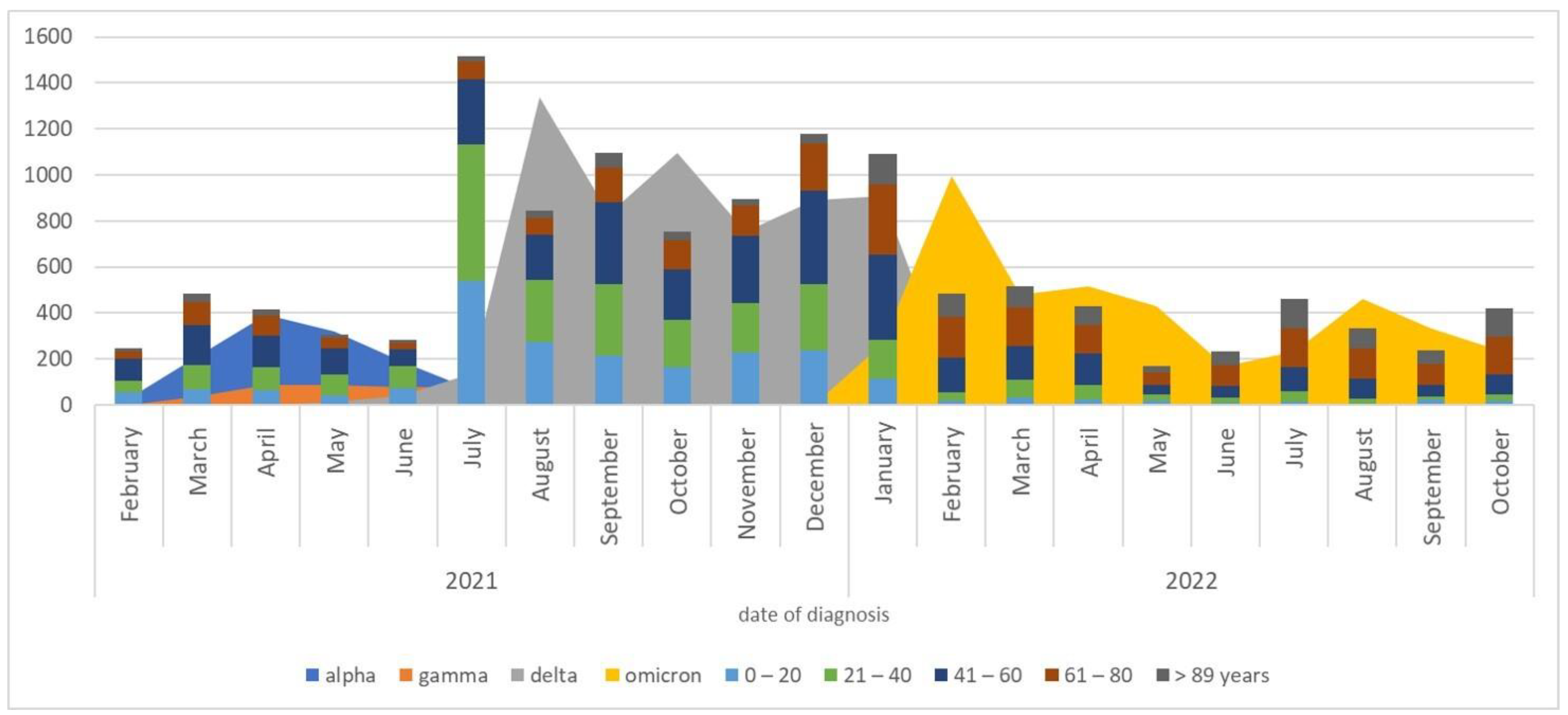
3.1. The Circulation of SARS-CoV-2 Variants
3.2. Circulation of Variants and Their Epidemiological Data
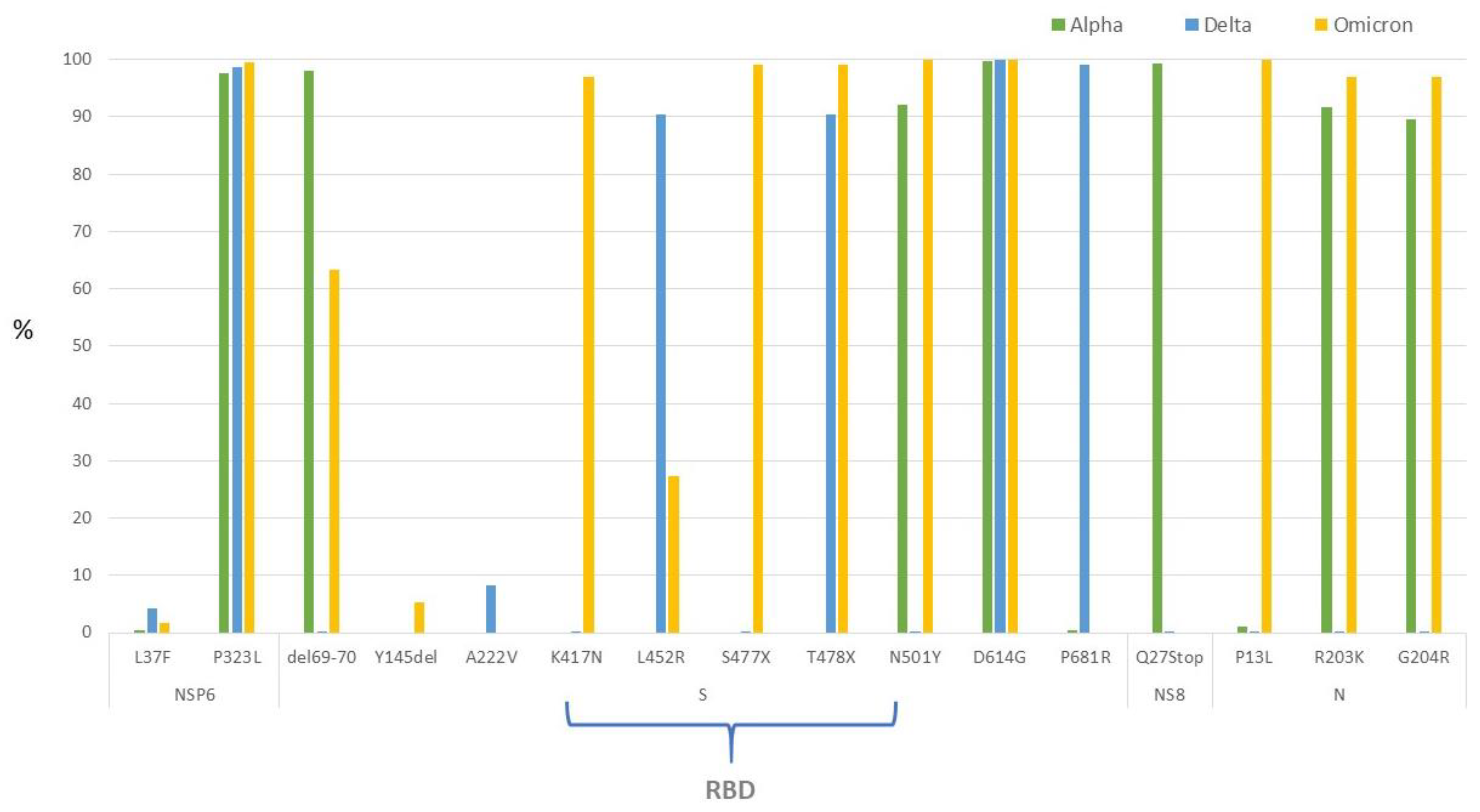
3.3. SARS-CoV-2 Genomic Characterization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. China Novel Coronavirus Investigating and Research Team. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 1 June 2022).
- Wang, S.; Xu, X.; Wei, C.; Li, S.; Zhao, J.; Zheng, Y.; Liu, X.; Zeng, X.; Yuan, W.; Peng, S. Molecular evolutionary characteristics of SARS-CoV-2 emerging in the United States. J. Med. Virol. 2022, 94, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Tosta, S.; Moreno, K.; Schuab, G.; Fonseca, V.; Segovia, F.M.C.; Kashima, S.; Elias, M.C.; Sampaio, S.C.; Ciccozzi, M.; Alcantara, L.C.J.; et al. Global SARS-CoV-2 genomic surveillance: What we have learned (so far). Infect. Genet. Evol. 2023, 108, 105405. [Google Scholar] [CrossRef]
- Scrima, M.; Cossu, A.M.; D’Andrea, E.L.; Bocchetti, M.; Abruzzese, Y.; Iannarone, C.; Miarelli, C.; Grisolia, P.; Melisi, F.; Genua, L.; et al. Genomic Characterization of the Emerging SARS-CoV-2 Lineage in Two Districts of Campania (Italy) Using Next-Generation Sequencing. Front. Virol. 2022, 2, 814114. [Google Scholar] [CrossRef]
- Bansal, K.; Kumar, S. Mutational cascade of SARS-CoV-2 leading to evolution and emergence of omicron variant. Virus Res. 2022, 315, 198765. [Google Scholar] [CrossRef] [PubMed]
- CDC Centers for Desease Control and Prevention; SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html#anchor_1633452601089t-classifications.html#anchor_1633452601089 (accessed on 1 August 2023).
- Konings, F.; Perkins, M.D.; Kuhn, J.H.; Pallen, M.J.; Alm, E.J.; Archer, B.N.; Barakat, A.; Bedford, T.; Bhiman, J.N.; Caly, L.; et al. SARS-CoV-2 Variants of Interest and Concern naming scheme conducive for global discourse. Nat. Microbiol. 2021, 6, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Duarte, C.M.; Ketcheson, D.I.; Eguíluz, V.M.; Agustí, S.; Fernández-Gracia, J.; Jamil, T.; Laiolo, E.; Gojobori, T.; Alam, I. Rapid evolution of SARS-CoV-2 challenges human defenses. Sci. Rep. 2022, 12, 6457. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Motayo, B.O.; Oluwasemowo, O.O.; Olusola, B.A.; Akinduti, P.A.; Arege, O.T.; Obafemi, Y.D.; Faneye, A.O.; Isibor, P.O.; Aworunse, O.S.; Oranusi, S.U. Evolution and genetic diversity of SARS-CoV-2 in Africa using whole genome sequences. Int. J. Infect. Dis. 2021, 103, 282–287. [Google Scholar] [CrossRef]
- Dhawan, M.; Saied, A.A.; Mitra, S.; Alhumaydhi, F.A.; Emran, T.B.; Wilairatana, P. Omicron variant (B.1.1.529) and its sublineages: What do we know so far amid the emergence of recombinant variants of SARS-CoV-2? Biomed. Pharmacother. 2022, 154, 113522. [Google Scholar] [CrossRef]
- Callaway, E. What Omicron’s BA.4 and BA.5 variants mean for the pandemic. Nature 2022, 606, 848–849. [Google Scholar] [CrossRef] [PubMed]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.E.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433. [Google Scholar] [CrossRef] [PubMed]
- WHO. Genomic Sequencing of SARS-CoV-2: A Guide to Implementation for Maximum Impact on Public Health, p. 94. Available online: https://www.who.int/publications/i/item/9789240018440 (accessed on 1 August 2023).
- European Centre for Disease Prevention and Control. Guidance for Representative and Targeted Genomic SARS-CoV-2 Monitoring—3 May 2021; ECDC: Stockholm, Sweden, 2021; Available online: https://www.ecdc.europa.eu/en/publications-data/guidance-representative-and-targeted-genomic-sars-cov-2-monitoring (accessed on 1 August 2023).
- Ministero della Salute. Indicazioni Operative Relative al Rischio di Diffusione di Nuove Varanti SARS-CoV-2 in Unione Europea/Spazio Economico Europeo (UE/SEE): Misure di Prevenzione per i Viaggiatori e Sorveglianza di Laboratorio. Available online: https://www.trovanorme.salute.gov.it/norme/renderNormsanPdf?anno-2021&codLeg-78153&parte-1%20&serie-null (accessed on 1 August 2023).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. NextStrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 13, 30494. [Google Scholar] [CrossRef] [PubMed]
- Istituto Superiore di Sanità. IRIDA-ARIES Platform. Available online: https://irida.iss.it/irida21-aries/login (accessed on 1 August 2023).
- ARTIC Network. Available online: https://artic.network/ncov-2019 (accessed on 19 June 2023).
- Rueca, M.; Giombini, E.; Messina, F.; Bartolini, B.; Di Caro, A.; Capobianchi, M.R.; Gruber, C.E. The Easy-to-Use SARS-CoV-2 Assembler for Genome Sequencing: Development Study. JMIR Bioinform. Biotech. 2022, 3, e31536. [Google Scholar] [CrossRef]
- Shepard, S.S.; Meno, S.; Bahl, J.; Wilson, M.M.; Barnes, J.; Neuhaus, E. Erratum to: Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genom. 2016, 17, 801. [Google Scholar] [CrossRef]
- De Sabato, L.; Vaccari, G.; Knijn, A.; Ianiro, G.; Di Bartolo, I.; Morabito, S. SARS-CoV-2 RECoVERY: A multi-platform open-source bioinformatic pipeline for the automatic construction and analysis of SARS-CoV-2 genomes from NGS sequencing data. bioRxiv 2021, 2021, 425365. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Della Ventura, C.; Menzo, S.; Bruzzone, B.; Sagradi, F.; Ceccherini-Silberstein, F.; Weisz, A.; Clementi, N.; Brindicci, G.; et al. Epidemiological and Clinical Features of SARS-CoV-2 Variants Circulating between April-December 2021 in Italy. Viruses. 2022, 14, 2508. [Google Scholar] [CrossRef]
- Regione Lazio. Determinazione n. G08714 del 30 Giugno 2021. “Realizzazione di una Rete di Laboratori Sub-Regionali per la Caratterizzazione dei Ceppi di SARS-CoV-2 Circolanti Nella Regione Lazio (WGSnet-Lazio)”. Bollettino Ufficiale della Regione Lazio dell’8 Luglio 2021, n. 68. Available online: https://sicer.regione.lazio.it/PublicBur/burlazio/FrontEnd# (accessed on 1 August 2023).
- Ministero della Salute. Relazione Tecnica Strategie di Sequenziamento per L’identificazione delle Varianti di SARS-CoV-2 ed il Monitoraggio della loro Circolazione in Italia—Indicazioni ad Interim. 2022. Available online: https://www.trovanorme.salute.gov.it/norme/renderNormsanPdf?anno=2022&codLeg=86233&parte=1%20&serie=null (accessed on 1 August 2023).
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev. Med. Virol. 2022, 32, e2381. [Google Scholar] [CrossRef]
- Ren, S.Y.; Wang, W.B.; Gao, R.D.; Zhou, A.M. Omicron variant (B.1.1.529) of SARS-CoV-2: Mutation, infectivity, transmission, and vaccine resistance. World J. Clin. Cases 2022, 10, 1–11. [Google Scholar] [CrossRef]
- Menni, C.; Valdes, A.M.; Polidori, L.; Antonelli, M.; Penamakuri, S.; Nogal, A.; Louca, P.; May, A.; Figueiredo, J.C.; Hu, C.; et al. Symptom prevalence, duration, and risk of hospital admission in individuals infected with SARS-CoV-2 during periods of omicron and delta variant dominance: A prospective observational study from the ZOE COVID Study. Lancet 2022, 399, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Chenchula, S.; Karunakaran, P.; Sharma, S.; Chavan, M. Current evidence on efficacy of COVID- 19 booster dose vaccination against the Omicron variant: A systematic review. J. Med. Virol. 2022, 94, 2969–2976. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Krüger, N.; Schulz, S.; Cossmann, A.; Rocha, C.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Moldenhauer, A.-S.; Winkler, M.S.; et al. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell 2022, 185, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Zhang, H.; Zhang, Y.; Lin, K.; Zhang, Y.; Wu, J.; Wan, Y.; Huang, Y.; Song, J.; Fu, Z.; et al. Omicron variant showed lower neutralizing sensitivity than other SARS-CoV-2 variants to immune sera elicited by vaccines after boost. Emerg. Microbes Infect. 2022, 11, 337–343. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Omicron Delta study group. Clinical progression, disease severity, and mortality among adults hospitalized with COVID-19 caused by the Omicron and Delta SARS-CoV-2 variants: A population-based, matched cohort study. PLoS ONE 2023, 18, e0282806. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mudi, S.R. Spike protein d614g and RDRP p323l: The SARS-CoV-2 mutations associated with severity of COVID-19. Genom. Inform. 2020, 18, e44. [Google Scholar] [CrossRef]
- Majumdar, P.; Niyogi, S. SARS-CoV-2 mutations: The biological trackway towards viral fitness. Epidemiol. Infect. 2021, 149, e110. [Google Scholar] [CrossRef]
- Di Giacomo, S.; Mercatelli, D.; Rakhimov, A.; Giorgi, F.M. Preliminary report on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike mutation T478K. J. Med. Virol. 2021, 93, 5638–5643. [Google Scholar] [CrossRef]
- Pondé, R.A.A. Physicochemical effect of the N501Y, E484K/Q, K417N/T, L452R and T478K mutations on the SARS-CoV-2 spike protein RBD and its influence on agent fitness and on attributes developed by emerging variants of concern. Virology 2022, 572, 44–54. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alpha | Gamma | Delta | Omicron | Total | ||
|---|---|---|---|---|---|---|
| (N = 1302) | (N = 460) | (N = 6113) | (N = 4543) | (N = 12418) | p Value | |
| Age, median (IQR) | 45, IQR (28–59) | 44, IQR (25–57) | 36, IQR (20–53) | 62, IQR (46–77) | 47, IQR (27–65) | 0.000 |
| Gender, n (%) | ||||||
| Female | 643 (49.4) | 232 (50.4) | 2972 (48.6) | 2262 (49.8) | 6109 (49.2) | |
| Male | 659 (50.6) | 228 (49.6) | 3141 (51.4) | 2281 (49.2) | 6309 (50.8) | 0.626 |
| Age groups in years, n (%) | ||||||
| 0–20 | 223 (17.1) | 81 (17.6) | 1648 (27.0) | 299 (6.6) | 2251 (18.1) | |
| 21–40 | 330 (25.4) | 125 (27.2) | 178 (29.1) | 586 (12.9) | 2821 (22.7) | |
| 41–60 | 445 (34.2) | 163 (35.4) | 1702 (27.8) | 1278 (28.1) | 3588 (28.9) | |
| 61–80 | 236 (18.1) | 62 (13.5) | 759 (12.4) | 1494 (39.9) | 2551 (20.5) | |
| >80 | 68 (5.2) | 29 (6.3) | 224 (3.7) | 886 (19.5) | 1207 (9.7) | 0.000 |
| Symptoms at diagnosis, n (%) | ||||||
| Yes | 777 (59.7) | 309 (67.2) | 3242 (53.0) | 1451 (31.9) | 5779 (46.5) | |
| No | 525 (40.3) | 151 (32.8) | 2871 (47.0) | 3092 (68.1) | 6639 (53.5) | 0.000 |
| Comorbidities, n (%) | ||||||
| Yes | 275 (21.1) | 91 (19.8) | 727 (11.9) | 755 (16.6) | 1848 (14.9) | |
| No | 537 (41.2) | 210 (45.7) | 2038 (33.3) | 213 (4.7) | 2998 (24.1) | |
| Missing data | 490 (37.6) | 159 (34.6) | 3348 (54.8) | 3575 (78.7) | 7572 (61) | 0.000 |
| Travel, n (%) | ||||||
| Yes | 60 (4.6) | 24 (5.2) | 491 (8) | 42 (0.9) | 617 (5) | |
| No | 813 (62.4) | 284 (61.7) | 2498 (40.9) | 802 (17.7) | 4397 (35.4) | |
| Missing data | 429 (32.9) | 152 (33) | 3124 (51.1) | 3699 (81.4) | 7404 (59.6) | 0.000 |
| Infection setting, n (%) | ||||||
| Healthcare setting | 32 (2.5) | 23 (5) | 60 (0.9) | 51 (1.2) | 166 (1.3) | |
| Household/friends | 414 (31.8) | 156 (33.9) | 1156 (18.9) | 141 (3.1) | 1867 (15) | |
| Other settings | 196 (15.1) | 58 (12.6) | 631 (10.3) | 70 (1.5) | 955 (7.7) | 0.000 |
| Missing data | 660 (50.7) | 223 (48.5) | 4266 (69.8) | 4281 (94.2) | 9430 (75.9) | |
| Vaccination status, n (%) | ||||||
| Not vaccinated | 1075 (82.6) | 334 (72.6) | 3428 (56.1) | 878 (19.3) | 5715 (46) | |
| Uncomplete vaccination | 110 (8.4) | 70 (15.2) | 491 (8) | 101 (2.2) | 772 (6.2) | |
| Fully vaccinated | 117 (9) | 56 (12.2) | 2194 (35.9) | 3564 (78.5) | 5931 (47.8) | 0.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rueca, M.; Berno, G.; Agresta, A.; Spaziante, M.; Gruber, C.E.M.; Fabeni, L.; Giombini, E.; Butera, O.; Barca, A.; Scognamiglio, P.; et al. Genomic and Epidemiologic Surveillance of SARS-CoV-2 in the Pandemic Period: Sequencing Network of the Lazio Region, Italy. Viruses 2023, 15, 2192. https://doi.org/10.3390/v15112192
Rueca M, Berno G, Agresta A, Spaziante M, Gruber CEM, Fabeni L, Giombini E, Butera O, Barca A, Scognamiglio P, et al. Genomic and Epidemiologic Surveillance of SARS-CoV-2 in the Pandemic Period: Sequencing Network of the Lazio Region, Italy. Viruses. 2023; 15(11):2192. https://doi.org/10.3390/v15112192
Chicago/Turabian StyleRueca, Martina, Giulia Berno, Alessandro Agresta, Martina Spaziante, Cesare Ernesto Maria Gruber, Lavinia Fabeni, Emanuela Giombini, Ornella Butera, Alessandra Barca, Paola Scognamiglio, and et al. 2023. "Genomic and Epidemiologic Surveillance of SARS-CoV-2 in the Pandemic Period: Sequencing Network of the Lazio Region, Italy" Viruses 15, no. 11: 2192. https://doi.org/10.3390/v15112192