

Exploring Tomato Fruit Viromes through Transcriptome Data Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Library Preparation
2.2. Data Processing and Transcriptome Assembly
2.3. Virus Identification
2.4. Analysis of Viral Proportion and Abundance
2.5. Investigation of Sound Treatment Effects on Tomato Viromes
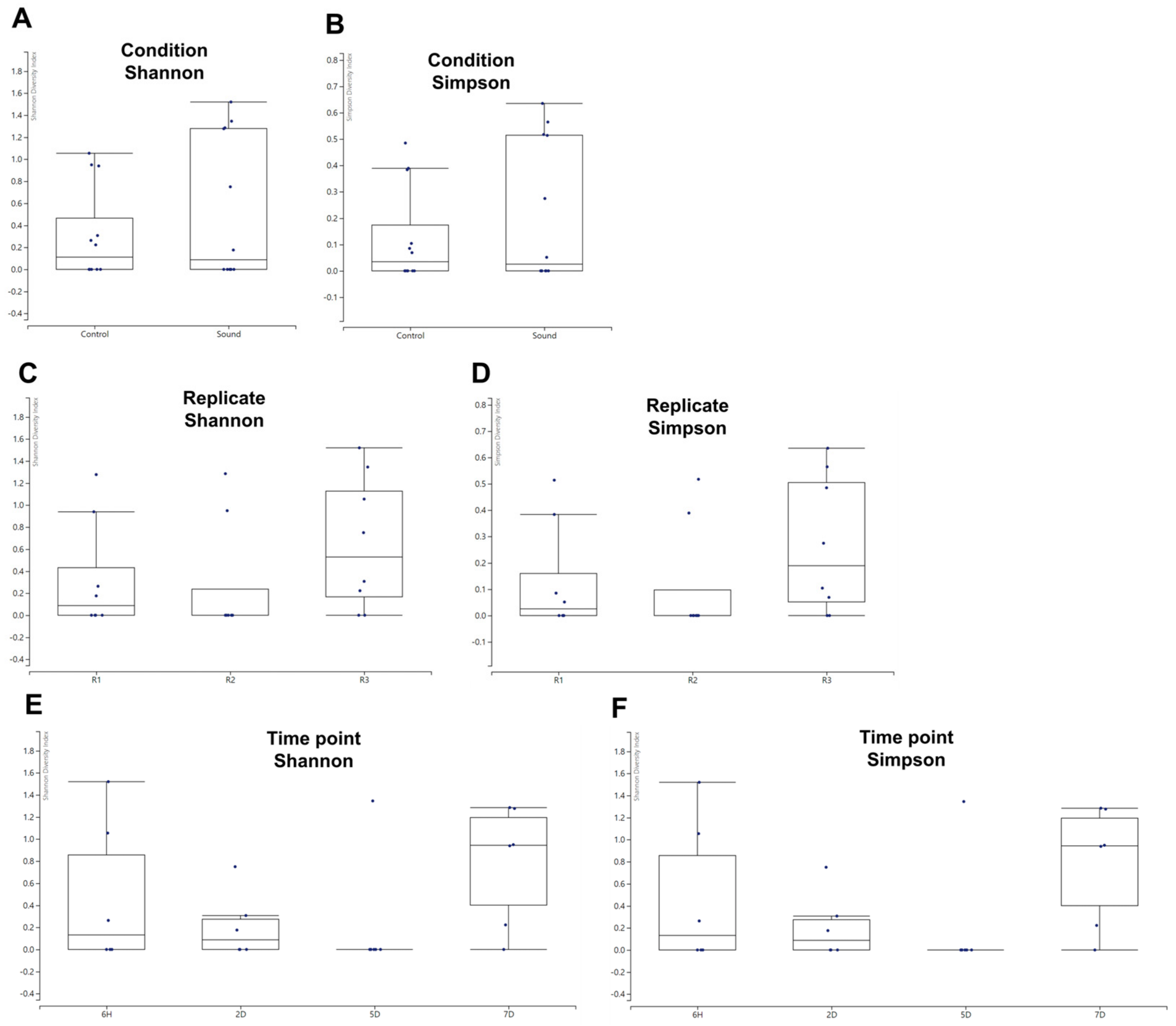
2.6. Alpha and Beta Diversity Analysis
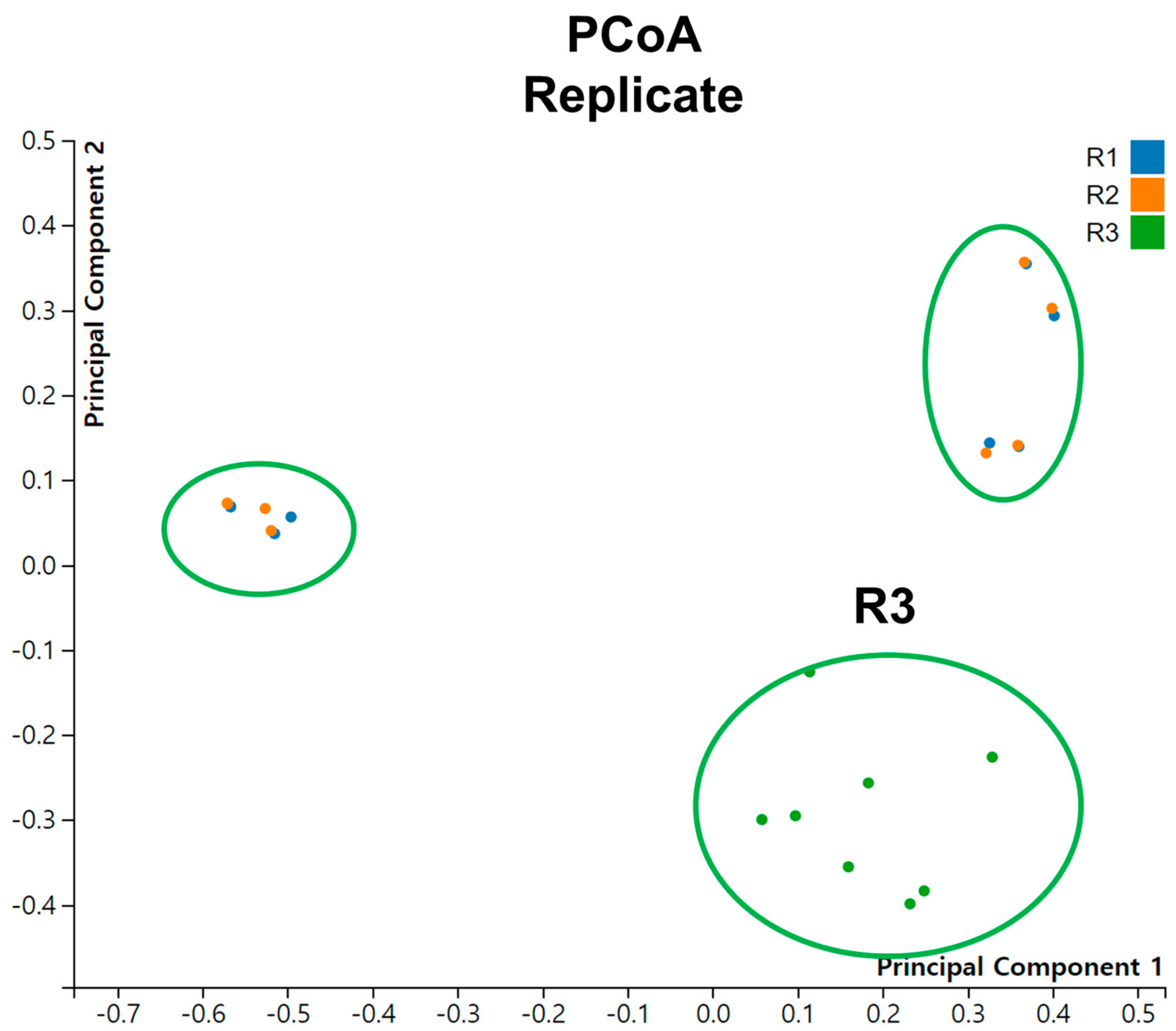
2.7. Principal Coordinate Analysis (PCoA)
2.8. Viral Genome Annotation
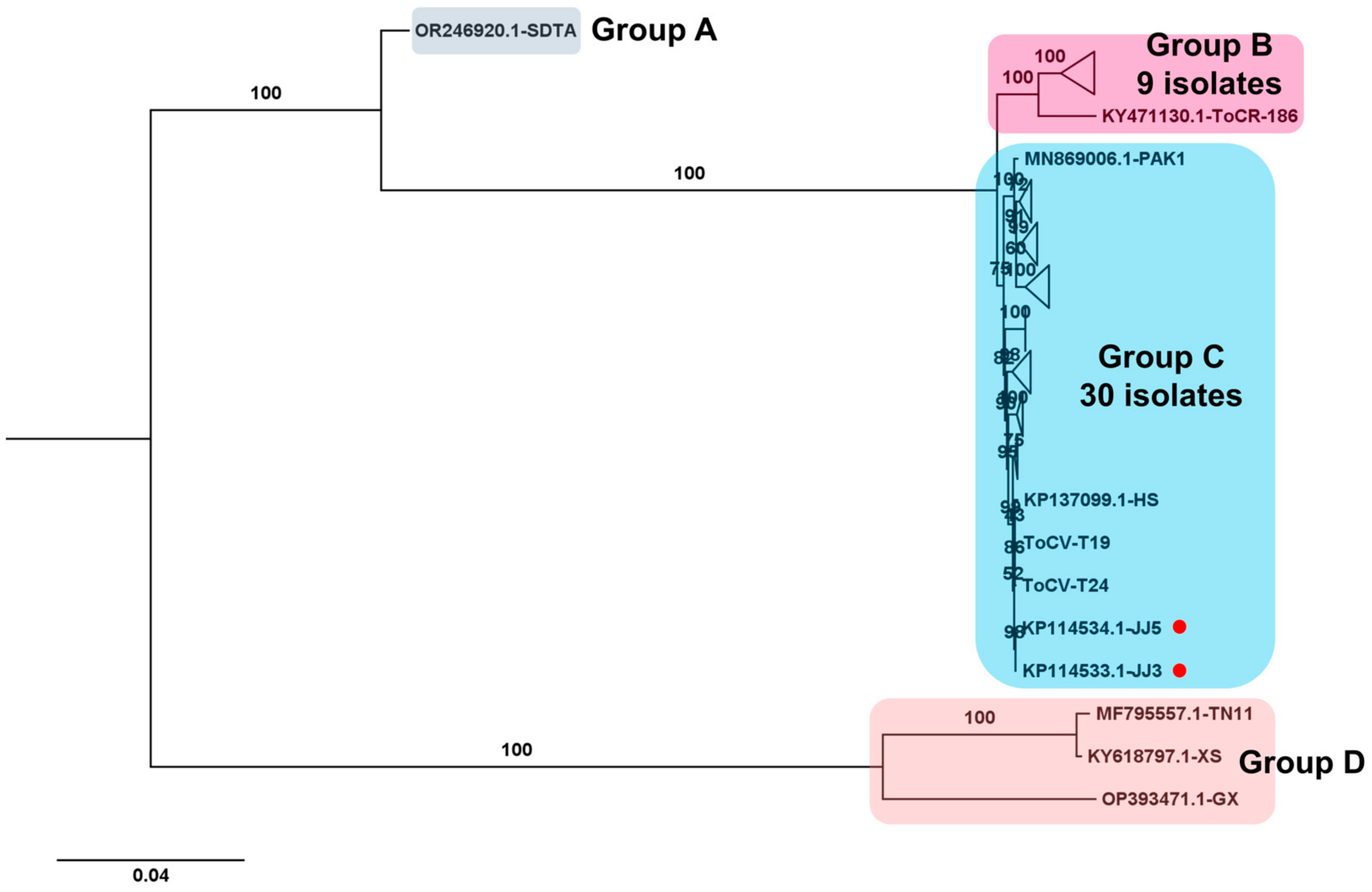
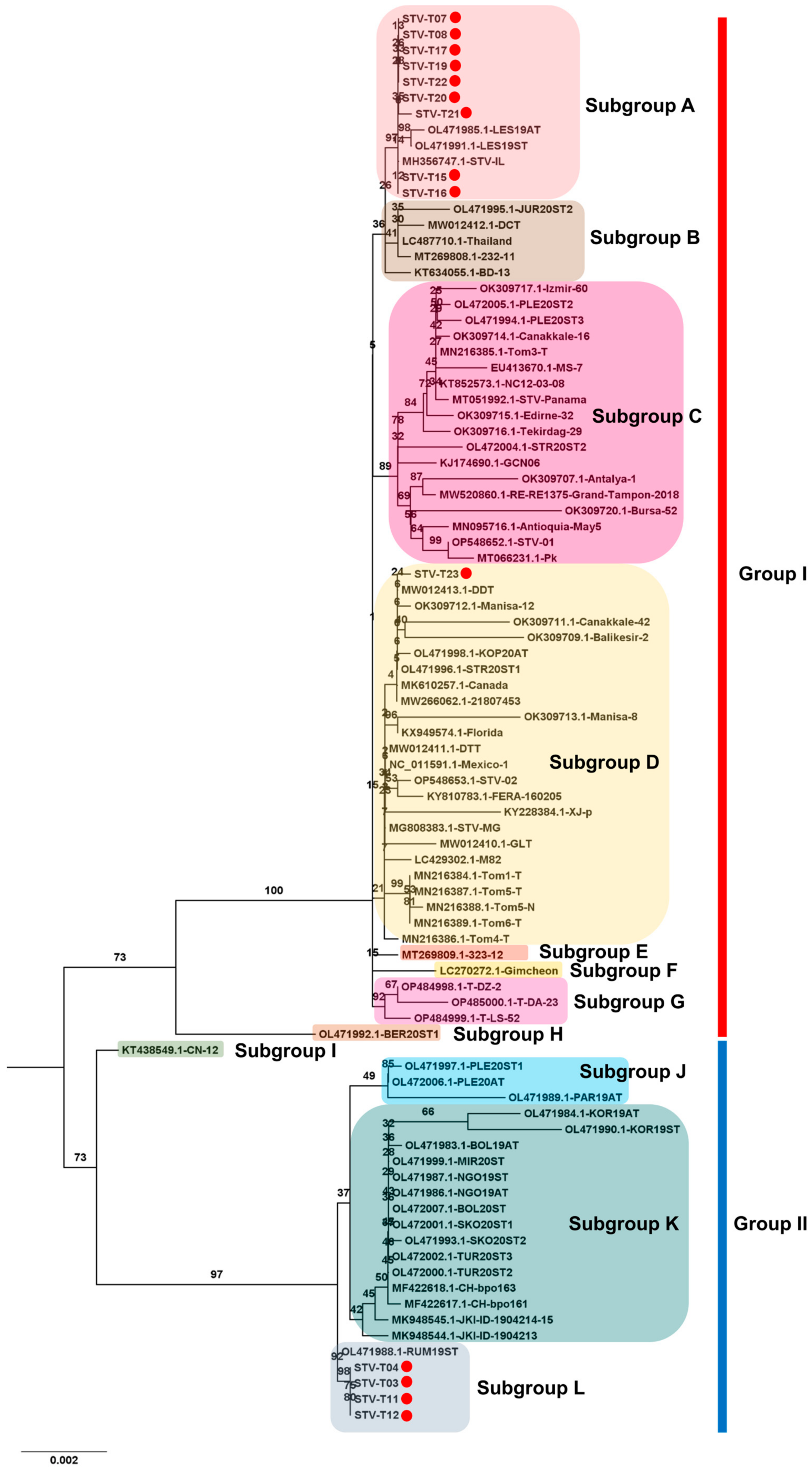
2.9. Phylogenetic Analysis
3. Results
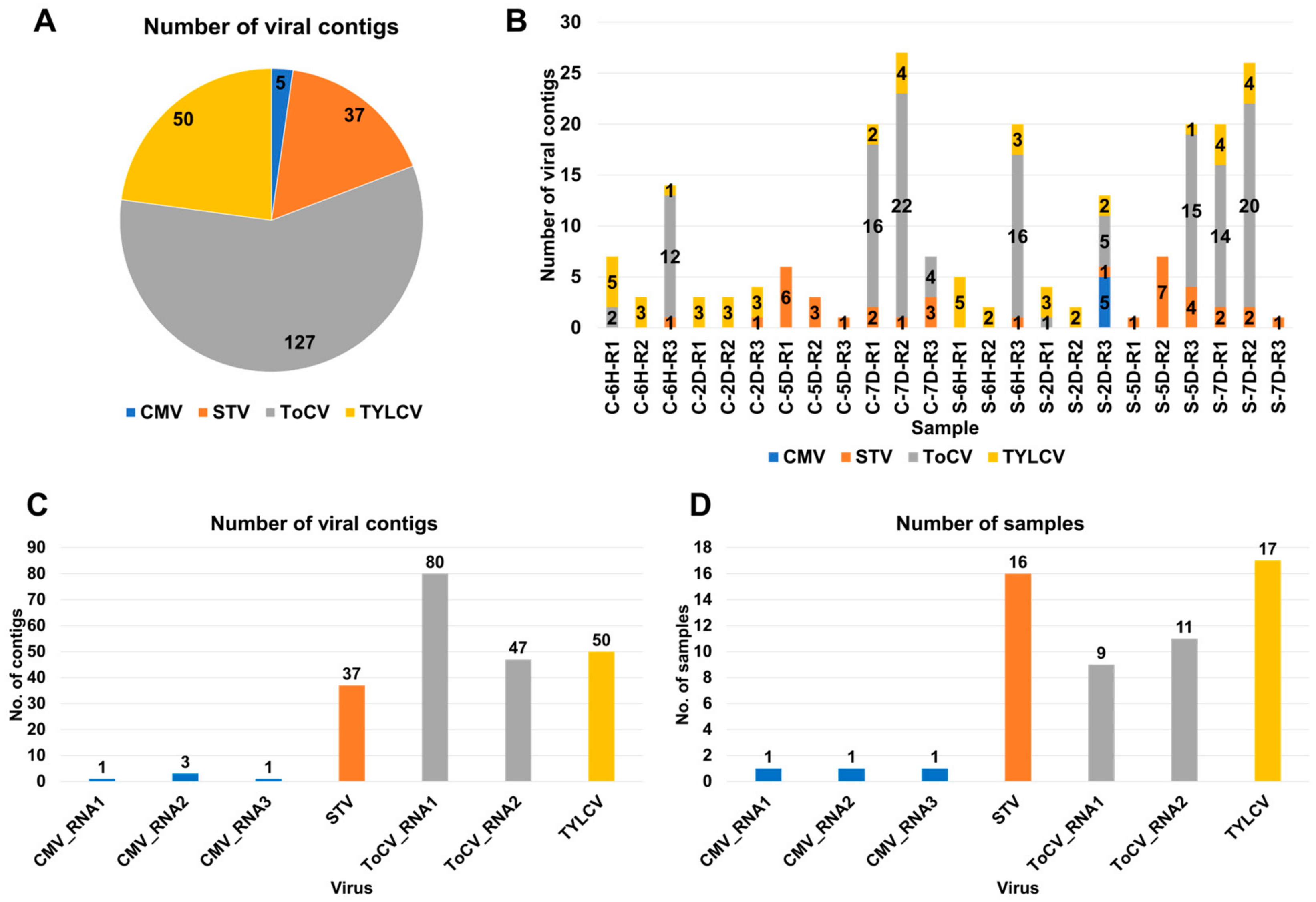
3.1. Viral Contig Identification and Genomic Classification
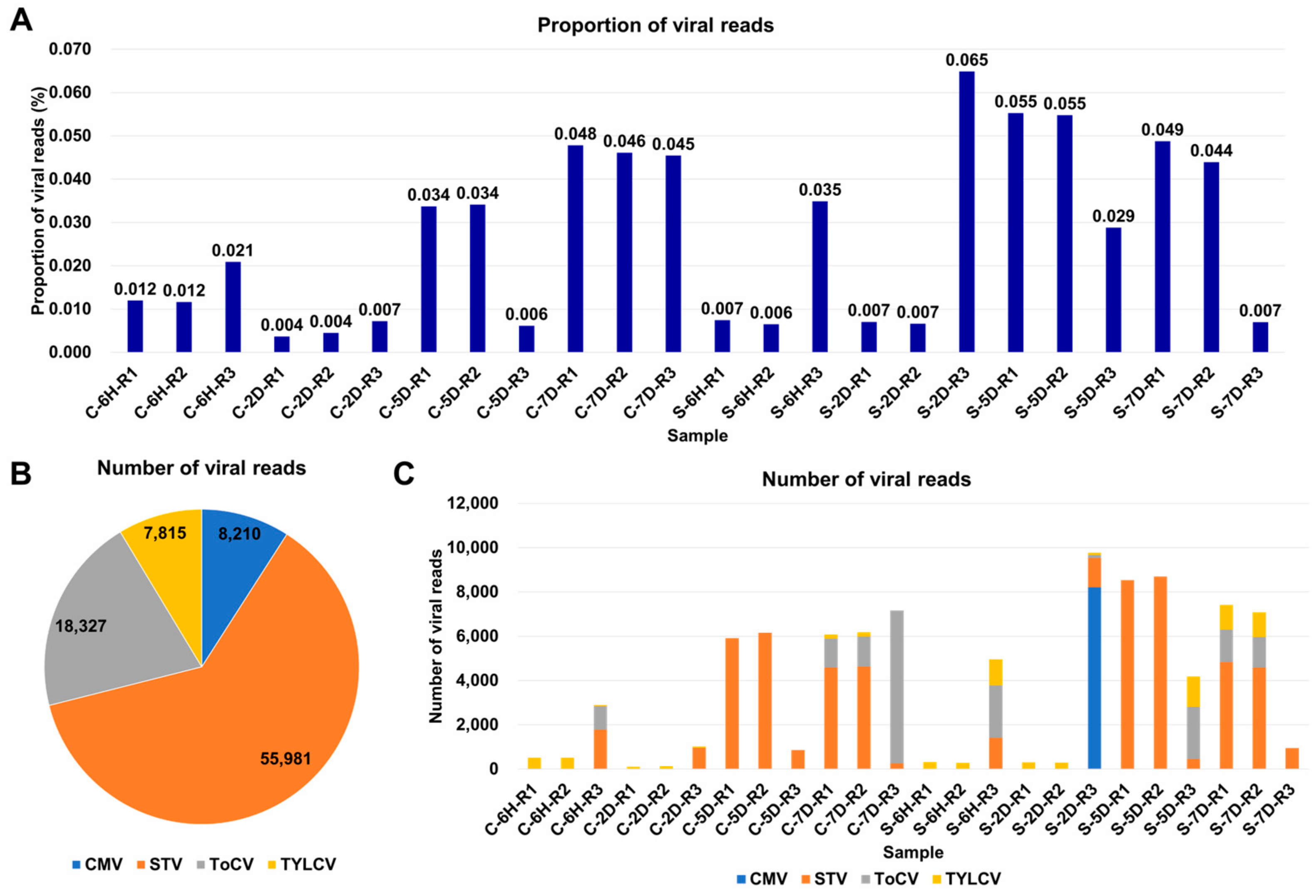
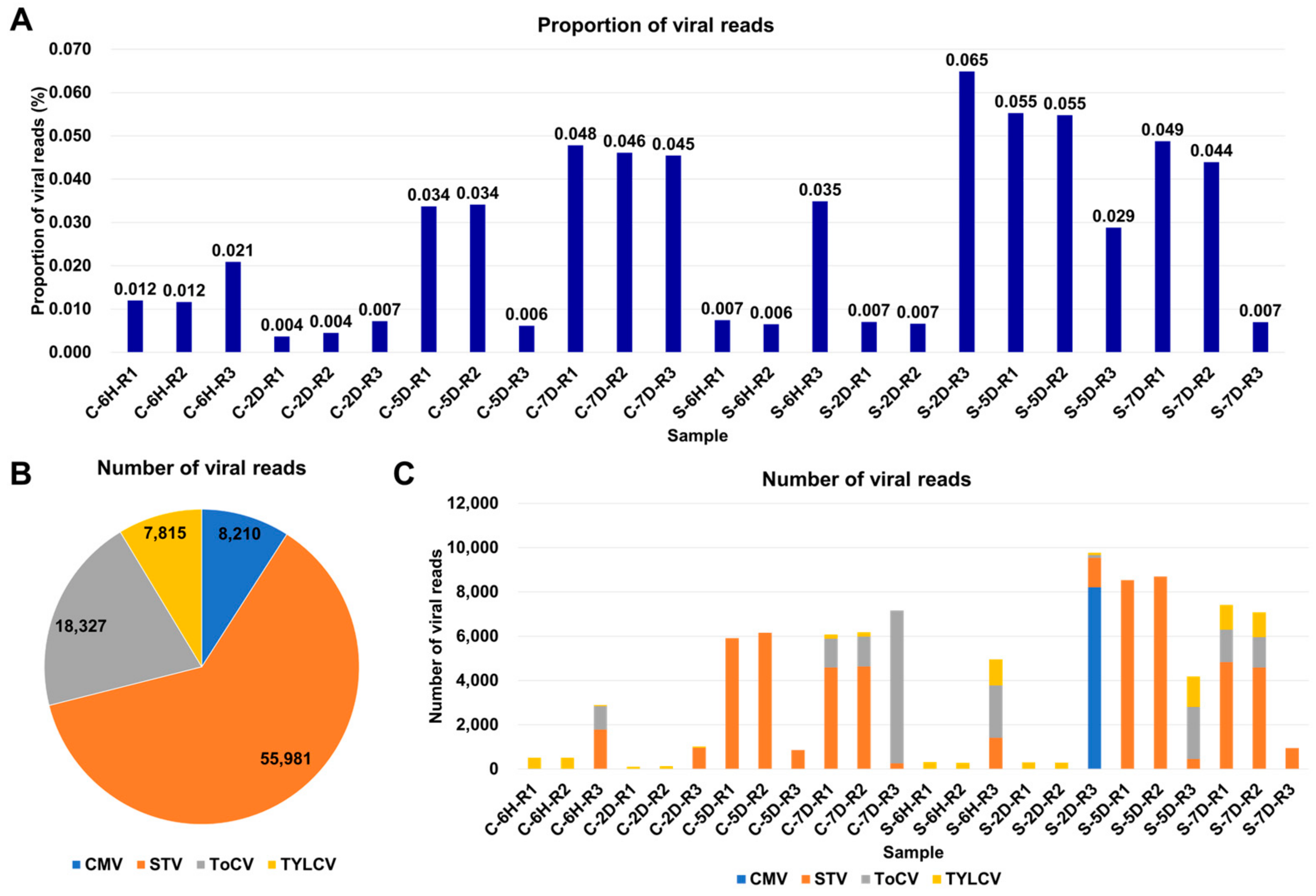
3.2. Viral-Read Proportions and Co-Infection Complexity
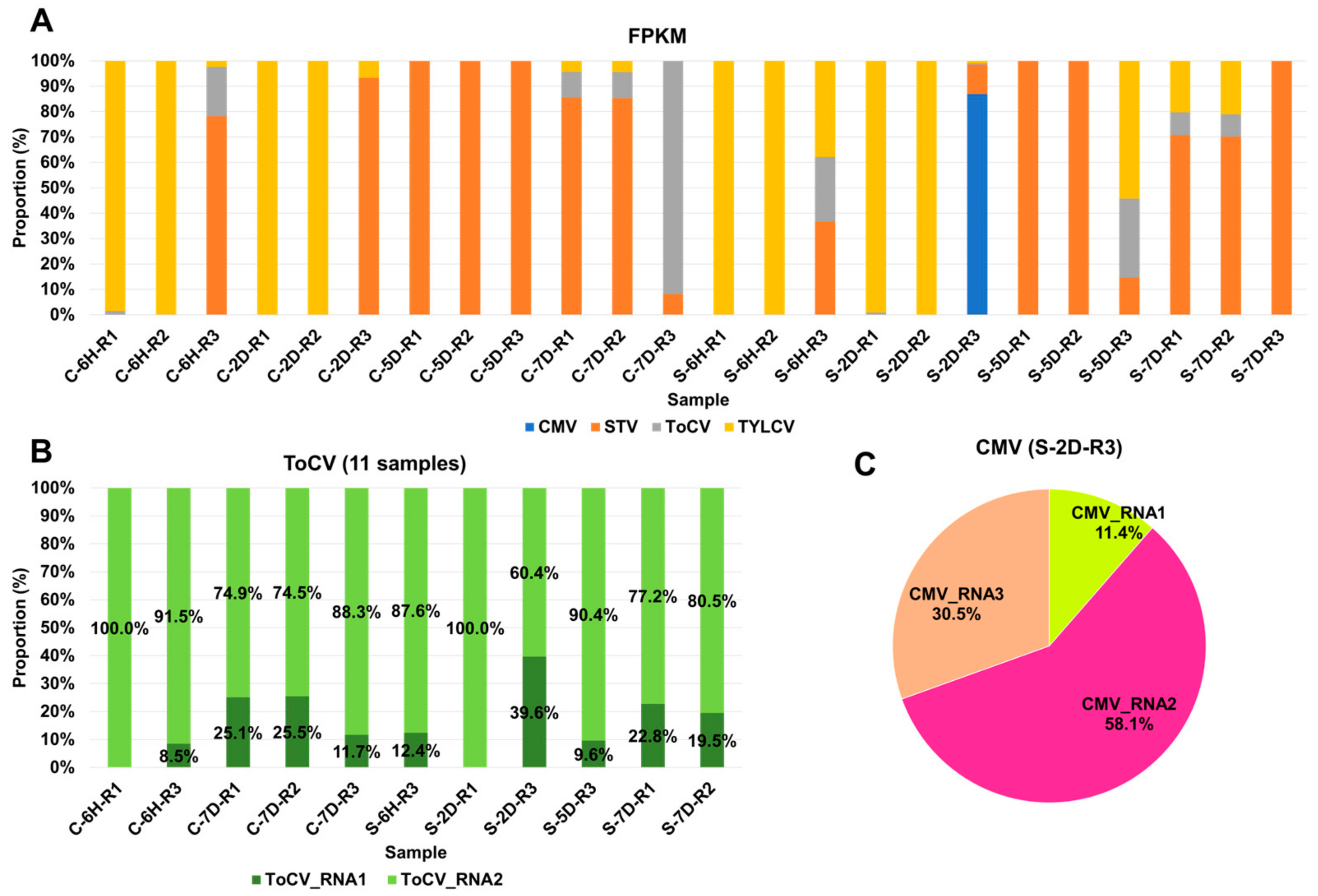
3.3. Viral Abundance and Genome Segment Analysis
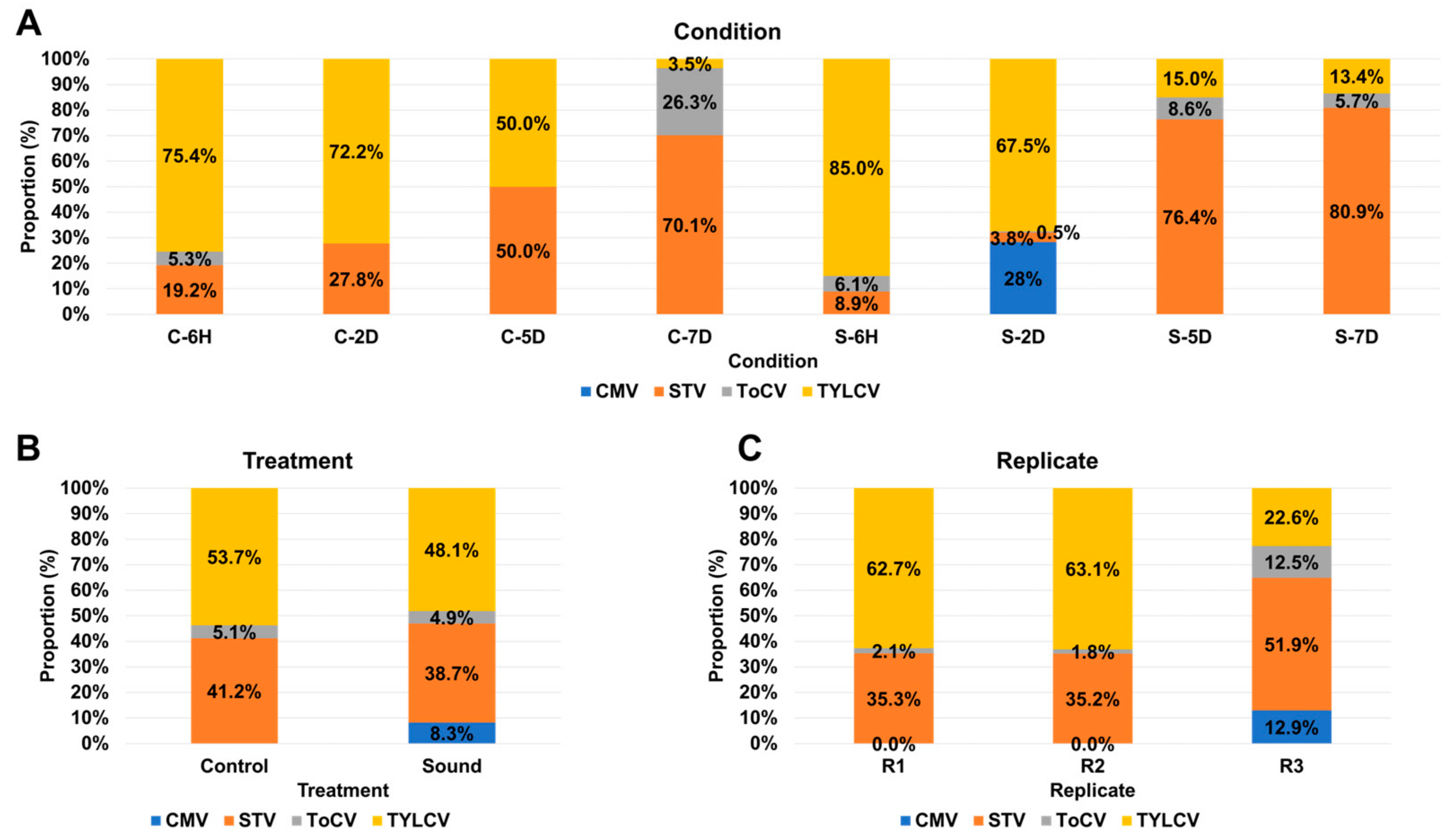
3.4. Impact of Sound Treatment on Viral Dynamics and Replication
3.5. Alpha and Beta Diversity Analysis of Viral Communities
3.6. Phylogenetic Analysis Using Obtained Viral Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dorais, M.; Ehret, D.L.; Papadopoulos, A.P. Tomato (Solanum lycopersicum) health components: From the seed to the consumer. Phytochem. Rev. 2008, 7, 231–250. [Google Scholar] [CrossRef]
- Egea, I.; Estrada, Y.; Flores, F.B.; Bolarín, M.C. Improving production and fruit quality of tomato under abiotic stress: Genes for the future of tomato breeding for a sustainable agriculture. Environ. Exp. Bot. 2022, 204, 105086. [Google Scholar] [CrossRef]
- Kissoudis, C.; Chowdhury, R.; van Heusden, S.; van de Wiel, C.; Finkers, R.; Visser, R.G.; Bai, Y.; van der Linden, G. Combined biotic and abiotic stress resistance in tomato. Euphytica 2015, 202, 317–332. [Google Scholar] [CrossRef]
- Mabvakure, B.; Martin, D.P.; Kraberger, S.; Cloete, L.; van Brunschot, S.; Geering, A.D.; Thomas, J.E.; Bananej, K.; Lett, J.-M.; Lefeuvre, P. Ongoing geographical spread of Tomato yellow leaf curl virus. Virology 2016, 498, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Marchant, W.G.; Mugerwa, H.; Gautam, S.; Al-Aqeel, H.; Polston, J.E.; Rennberger, G.; Smith, H.; Turechek, B.; Adkins, S.; Brown, J.K. Phylogenomic and population genetics analyses of extant tomato yellow leaf curl virus strains on a global scale. Front. Virol. 2023, 3, 1221156. [Google Scholar] [CrossRef]
- Roselló, S.; Díez, M.J.; Nuez, F. Viral diseases causing the greatest economic losses to the tomato crop. I. The Tomato spotted wilt virus—A review. Sci. Hortic. 1996, 67, 117–150. [Google Scholar] [CrossRef]
- Dhaliwal, M.; Jindal, S.; Sharma, A.; Prasanna, H. Tomato yellow leaf curl virus disease of tomato and its management through resistance breeding: A review. J. Hortic. Sci. Biotechnol. 2020, 95, 425–444. [Google Scholar] [CrossRef]
- Saidi, M.; Warade, S.D. Tomato breeding for resistance to Tomato spotted wilt virus (TSWV): An overview of conventional and molecular approaches. Czech J. Genet. Plant Breed. 2008, 44, 83–92. [Google Scholar] [CrossRef]
- Villamor, D.; Ho, T.; Al Rwahnih, M.; Martin, R.; Tzanetakis, I. High throughput sequencing for plant virus detection and discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef]
- Rivarez, M.P.S.; Vučurović, A.; Mehle, N.; Ravnikar, M.; Kutnjak, D. Global advances in tomato virome research: Current status and the impact of high-throughput sequencing. Front. Microbiol. 2021, 12, 671925. [Google Scholar] [CrossRef]
- Xu, C.; Sun, X.; Taylor, A.; Jiao, C.; Xu, Y.; Cai, X.; Wang, X.; Ge, C.; Pan, G.; Wang, Q. Diversity, distribution, and evolution of tomato viruses in China uncovered by small RNA sequencing. J. Virol. 2017, 91, e00173-17. [Google Scholar] [CrossRef]
- Rivarez, M.P.S.; Pecman, A.; Bačnik, K.; Maksimović, O.; Vučurović, A.; Seljak, G.; Mehle, N.; Gutiérrez-Aguirre, I.; Ravnikar, M.; Kutnjak, D. In-depth study of tomato and weed viromes reveals undiscovered plant virus diversity in an agroecosystem. Microbiome 2023, 11, 60. [Google Scholar] [CrossRef]
- Ma, Y.; Marais, A.; Lefebvre, M.; Faure, C.; Candresse, T. Metagenomic analysis of virome cross-talk between cultivated Solanum lycopersicum and wild Solanum nigrum. Virology 2020, 540, 38–44. [Google Scholar] [CrossRef]
- Dias, N.P.; Hu, R.; Hale, F.A.; Hansen, Z.R.; Wszelaki, A.; Domier, L.L.; Hajimorad, M. Viromes of Field-Grown Tomatoes and Peppers in Tennessee Revealed by RNA Sequencing Followed by Bioinformatic Analysis. Plant Health Prog. 2023, 24, 207–213. [Google Scholar] [CrossRef]
- Shin, J.-S.; Han, J.-H.; Shin, Y.-J.; Kwak, H.-R.; Choi, H.-S.; Kim, J.-S. Specific primer sets for RT-PCR detection of major RNA viruses of tomato plants in Korea. Res. Plant Dis. 2017, 23, 193–201. [Google Scholar] [CrossRef]
- Kim, S.H.; Oh, S.; Oh, T.-K.; Park, J.S.; Kim, S.C.; Kim, S.H.; Kim, Y.S.; Hong, J.K.; Sim, S.-Y.; Park, K.S. Genetic diversity of tomato-infecting Tomato yellow leaf curl virus (TYLCV) isolates in Korea. Virus Genes 2011, 42, 117–127. [Google Scholar] [CrossRef]
- Oh, J.; Lee, H.-K.; Park, C.-Y.; Yeom, Y.-A.; Min, H.-G.; Yang, H.-J.; Jeong, R.-D.; Kim, H.; Moon, J.-S.; Lee, S.-H. First report of Southern tomato virus in tomato (Solanum lycopersicum) in Korea. Plant Dis. 2018, 102, 1467. [Google Scholar] [CrossRef]
- Kim, M.K.; Kwak, H.R.; Han, J.H.; Ko, S.J.; Lee, S.H.; Park, J.W.; Jonson, M.G.; Kim, K.H.; Kim, J.S.; Choi, H.S. Isolation and characterization of Pepper mottle virus infecting tomato in Korea. Plant Pathol. J. 2008, 24, 152–158. [Google Scholar] [CrossRef]
- Kim, M.-K.; Kwak, H.-R.; Jeong, S.-G.; Ko, S.-J.; Lee, S.-H.; Park, J.-W.; Kim, K.-H.; Choi, H.-S.; Cha, B.-J. First report on Tomato bushy stunt virus infecting tomato in Korea. Plant Pathol. J. 2007, 23, 143–150. [Google Scholar] [CrossRef]
- Choi, G.-W.; Kim, B.; Ju, H.; Cho, S.; Seo, E.; Kim, J.; Park, J.; Hammond, J.; Lim, H.-S. Dual infections of Tomato mosaic virus (ToMV) and Tomato yellow leaf curl virus (TYLCV), or Tomato mosaic virus (ToMV) and Tomato chlorosis virus (ToCV), detected in tomato fields located in Chungcheongnam-do in 2017. Korean J. Agric. Sci. 2018, 45, 38–42. [Google Scholar]
- Choi, H.; Jo, Y.; Chung, H.; Choi, S.Y.; Kim, S.-M.; Hong, J.-S.; Lee, B.C.; Cho, W.K. Investigating Variability in Viral Presence and Abundance across Soybean Seed Development Stages Using Transcriptome Analysis. Plants 2023, 12, 3257. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Jo, Y.; Cho, W.K. In Silico Virome Analysis of Chinese Narcissus Transcriptomes Reveals Diverse Virus Species and Genetic Diversity at Different Flower Development Stages. Biology 2023, 12, 1094. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Choi, H.; Chu, H.; Cho, W.K. Unveiling Mycoviromes Using Fungal Transcriptomes. Int. J. Mol. Sci. 2022, 23, 10926. [Google Scholar] [CrossRef]
- Jo, Y.; Back, C.-G.; Kim, K.-H.; Chu, H.; Lee, J.H.; Moh, S.H.; Cho, W.K. Using RNA-sequencing data to examine tissue-specific garlic microbiomes. Int. J. Mol. Sci. 2021, 22, 6791. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, S.-K.; Jung, J.; Jeong, M.-J.; Ryu, C.-M. Exploring the sound-modulated delay in tomato ripening through expression analysis of coding and non-coding RNAs. Ann. Bot. 2018, 122, 1231–1244. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Jin, B.T.; Xu, F.; Ng, R.T.; Hogg, J.C. Mian: Interactive web-based microbiome data table visualization and machine learning platform. Bioinformatics 2022, 38, 1176–1178. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Kil, E.-J.; Kwak, H.-R.; Kim, M.; Seo, J.-K.; Lee, S.; Choi, H.-S. Phylogenetic characterization of Tomato chlorosis virus population in Korea: Evidence of reassortment between isolates from different origins. Plant Pathol. J. 2018, 34, 199. [Google Scholar] [CrossRef]
- Fortes, I.M.; Fernández-Muñoz, R.; Moriones, E. The crinivirus tomato chlorosis virus compromises the control of tomato yellow leaf curl virus in tomato plants by the Ty-1 gene. Phytopathology 2023, 113, 1347–1359. [Google Scholar] [CrossRef]
- Kumar, M.; Kavalappara, S.R.; McAvoy, T.; Hutton, S.; Simmons, A.M.; Bag, S. Association of Tomato Chlorosis Virus Complicates the Management of Tomato Yellow Leaf Curl Virus in Cultivated Tomato (Solanum lycopersicum) in the Southern United States. Horticulturae 2023, 9, 948. [Google Scholar] [CrossRef]
- Elvira González, L.; Peiró, R.; Rubio, L.; Galipienso, L. Persistent southern tomato virus (STV) interacts with cucumber mosaic and/or pepino mosaic virus in mixed-infections modifying plant symptoms, viral titer and small RNA accumulation. Microorganisms 2021, 9, 689. [Google Scholar] [CrossRef]
- Wintermantel, W.M.; Wisler, G.C. Vector specificity, host range, and genetic diversity of Tomato chlorosis virus. Plant Dis. 2006, 90, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Sharma, N.; Hari-Gowthem, G.; Muthamilarasan, M.; Prasad, M. Tomato yellow leaf curl virus: Impact, challenges, and management. Trends Plant Sci. 2020, 25, 897–911. [Google Scholar] [CrossRef]
- Skelton, A.; van Gemert, J.; Fowkes, A.; Frew, L.; Alraiss, K.; Hodgson, R.; Cressey, J.; Barnhoorn, R.; Macarthur, R.; Stijger, I. Detection of tomato brown rugose fruit virus is influenced by infection at different growth stages and sampling from different plant parts. Plant Pathol. 2023, 72, 1491–1504. [Google Scholar] [CrossRef]
- Sabanadzovic, S.; Valverde, R.A.; Brown, J.K.; Martin, R.R.; Tzanetakis, I.E. Southern tomato virus: The link between the families Totiviridae and Partitiviridae. Virus Res. 2009, 140, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Weaver, S.C.; Winegar, R.A. Legal, technical, and interpretational considerations in the forensic analysis of viruses. J. Forensic Sci. 2013, 58, 344–357. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Library Name | Sample Name | Condition | Time Point | Replicate | Acc. No. |
|---|---|---|---|---|---|---|
| 1 | T13 | C-6H-R1 | Control | 6H | R1 | SRR6234985 |
| 2 | T14 | C-6H-R2 | Control | 6H | R2 | SRR6234986 |
| 3 | T23 | C-6H-R3 | Control | 6H | R3 | SRR7668114 |
| 4 | T09 | C-2D-R1 | Control | 2D | R1 | SRR6234981 |
| 5 | T10 | C-2D-R2 | Control | 2D | R2 | SRR6234982 |
| 6 | T21 | C-2D-R3 | Control | 2D | R3 | SRR7668112 |
| 7 | T11 | C-5D-R1 | Control | 5D | R1 | SRR6234983 |
| 8 | T12 | C-5D-R2 | Control | 5D | R2 | SRR6234984 |
| 9 | T22 | C-5D-R3 | Control | 5D | R3 | SRR7668113 |
| 10 | T15 | C-7D-R1 | Control | 7D | R1 | SRR6234987 |
| 11 | T16 | C-7D-R2 | Control | 7D | R2 | SRR6234988 |
| 12 | T24 | C-7D-R3 | Control | 7D | R3 | SRR7668115 |
| 13 | T05 | S-6H-R1 | Sound | 6H | R1 | SRR6234977 |
| 14 | T06 | S-6H-R2 | Sound | 6H | R2 | SRR6234978 |
| 15 | T19 | S-6H-R3 | Sound | 6H | R3 | SRR7668110 |
| 16 | T01 | S-2D-R1 | Sound | 2D | R1 | SRR6234973 |
| 17 | T02 | S-2D-R2 | Sound | 2D | R2 | SRR6234974 |
| 18 | T17 | S-2D-R3 | Sound | 2D | R3 | SRR7668108 |
| 19 | T03 | S-5D-R1 | Sound | 5D | R1 | SRR6234975 |
| 20 | T04 | S-5D-R2 | Sound | 5D | R2 | SRR6234976 |
| 21 | T18 | S-5D-R3 | Sound | 5D | R3 | SRR7668109 |
| 22 | T07 | S-7D-R1 | Sound | 7D | R1 | SRR6234979 |
| 23 | T08 | S-7D-R2 | Sound | 7D | R2 | SRR6234980 |
| 24 | T20 | S-7D-R3 | Sound | 7D | R3 | SRR7668111 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, Y.; Choi, H.; Lee, B.C.; Hong, J.-S.; Kim, S.-M.; Cho, W.K. Exploring Tomato Fruit Viromes through Transcriptome Data Analysis. Viruses 2023, 15, 2139. https://doi.org/10.3390/v15112139
Jo Y, Choi H, Lee BC, Hong J-S, Kim S-M, Cho WK. Exploring Tomato Fruit Viromes through Transcriptome Data Analysis. Viruses. 2023; 15(11):2139. https://doi.org/10.3390/v15112139
Chicago/Turabian StyleJo, Yeonhwa, Hoseong Choi, Bong Choon Lee, Jin-Sung Hong, Sang-Min Kim, and Won Kyong Cho. 2023. "Exploring Tomato Fruit Viromes through Transcriptome Data Analysis" Viruses 15, no. 11: 2139. https://doi.org/10.3390/v15112139
APA StyleJo, Y., Choi, H., Lee, B. C., Hong, J.-S., Kim, S.-M., & Cho, W. K. (2023). Exploring Tomato Fruit Viromes through Transcriptome Data Analysis. Viruses, 15(11), 2139. https://doi.org/10.3390/v15112139