
Evaluation of Direct Detection Protocols for Poliovirus from Stool Samples of Acute Flaccid Paralysis Patients


Abstract
:1. Introduction
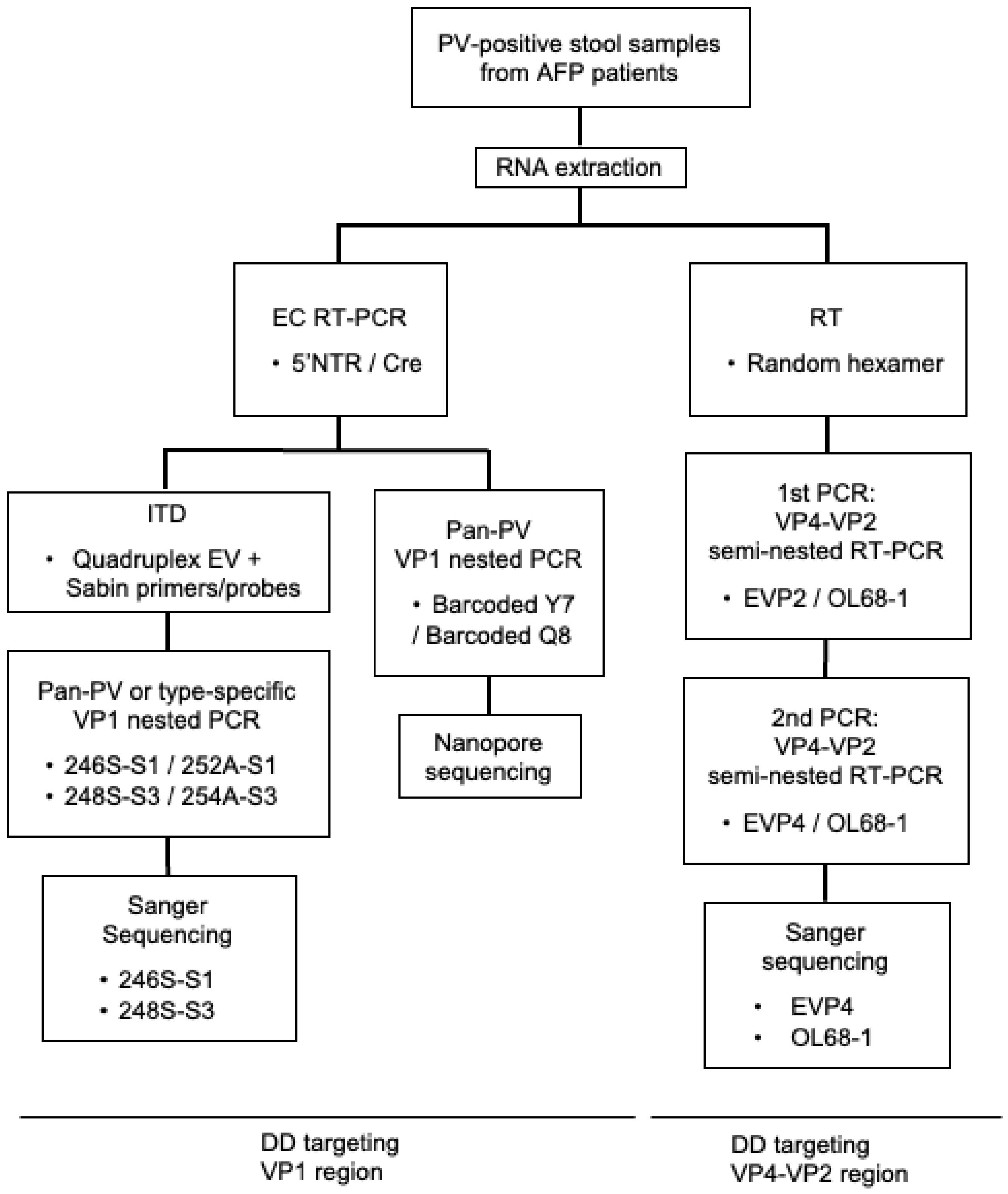
2. Materials and Methods
3. Results
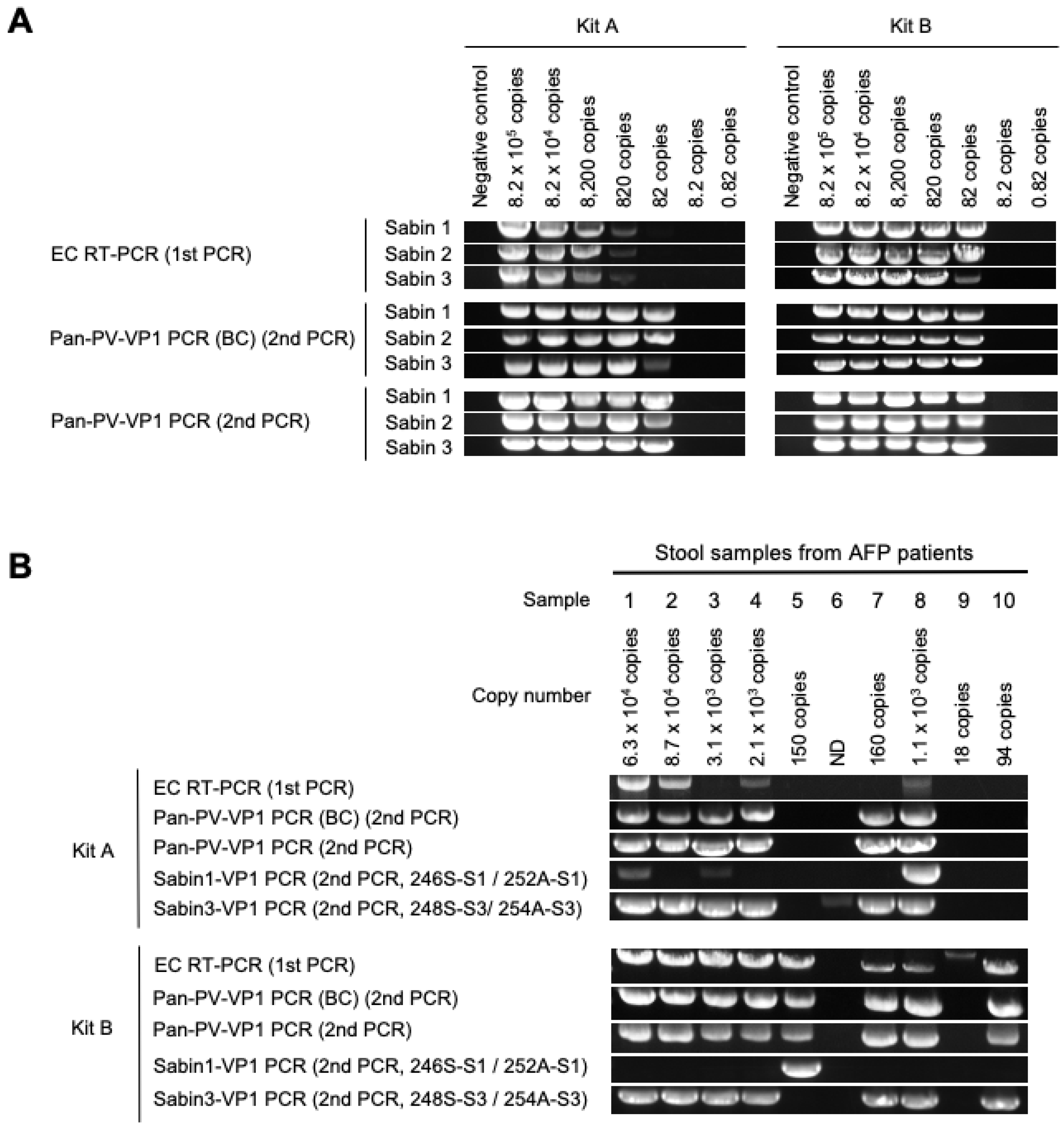
3.1. DD Targeting the VP1 Region
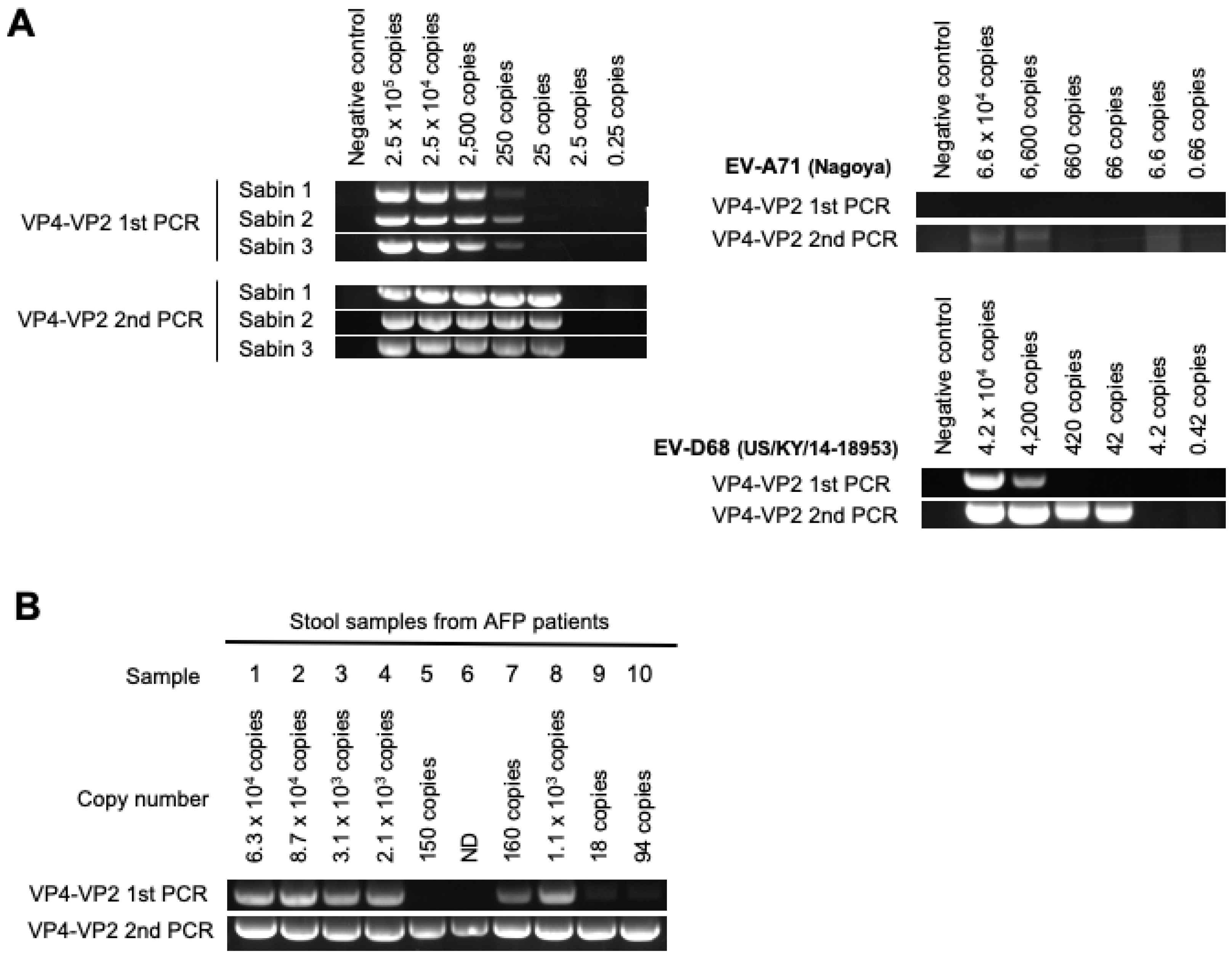
3.2. DD Targeting the VP4–VP2 Region
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Polio Eradication Strategy 2022–2026. 2021. Available online: https://polioeradication.org/wp-content/uploads/2021/10/9789240031937-eng.pdf (accessed on 30 August 2023).
- World Health Organization. Global Polio Surveillance Action Plan 2022–2024. 2022. Available online: https://polioeradication.org/wp-content/uploads/2022/05/GPSAP-2022-2024-EN.pdf (accessed on 30 August 2023).
- World Health Organization. Poliovirus. Laboratory. Manual, 5th ed.; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Duizer, E.; Ruijs, W.L.; Putri Hintaran, A.D.; Hafkamp, M.C.; van der Veer, M.; Te Wierik, M.J. Wild poliovirus type 3 (WPV3)-shedding event following detection in environmental surveillance of poliovirus essential facilities. Euro. Surveill. 2023, 28, 2300049. [Google Scholar]
- Arita, M.; Kilpatrick, D.R.; Nakamura, T.; Burns, C.C.; Bukbuk, D.; Oderinde, S.B.; Oberste, M.S.; Kew, O.M.; Pallansch, M.A.; Shimizu, H. Development of an Efficient Entire-Capsid-Coding-Region Amplification Method for Direct Detection of Poliovirus from Stool Extracts. J. Clin. Microbiol. 2015, 53, 73–78. [Google Scholar] [CrossRef]
- Shaw, A.G.; Majumdar, M.; Troman, C.; O’Toole, A.; Benny, B.; Abraham, D.; Praharaj, I. Rapid and Sensitive Direct Detection and Identification of Poliovirus from Stool and Environmental Surveillance Samples by Use of Nanopore Sequencing. J. Clin. Microbiol. 2020, 58, e00920-20. [Google Scholar] [CrossRef]
- Harvala, H.; Broberg, E.; Benschop, K.; Berginc, N.; Ladhani, S.; Susi, P.; Christiansen, C.; McKenna, J.; Allen, D.; Makiello, P.; et al. Recommendations for enterovirus diagnostics and characterisation within and beyond Europe. J. Clin. Virol. 2018, 101, 11–17. [Google Scholar] [CrossRef]
- Ishiko, H.; Shimada, Y.; Yonaha, M.; Hashimoto, O.; Hayashi, A.; Sakae, K.; Takeda, N. Molecular diagnosis of human enteroviruses by phylogeny-based classification by use of the VP4 sequence. J. Infect. Dis. 2002, 185, 744–754. [Google Scholar] [CrossRef]
- Perera, D.; Shimizu, H.; Yoshida, H.; Van Tu, P.; Ishiko, H.; McMinn, P.C.; Cardosa, M.J. A comparison of the VP1, VP2, and VP4 regions for molecular typing of human enteroviruses. J. Med. Virol. 2010, 82, 649–657. [Google Scholar] [CrossRef]
- Zhou, Y.; Qiu, Q.; Luo, K.; Liao, Q.; Li, Y.; Cui, P.; Liang, L.; Cheng, Y.; Wang, L.; Wang, K.; et al. Molecular strategy for the direct detection and identification of human enteroviruses in clinical specimens associated with hand, foot and mouth disease. PLoS ONE 2020, 15, e0241614. [Google Scholar] [CrossRef]
- Kew, O.; Morris-Glasgow, V.; Landaverde, M.; Burns, C.; Shaw, J.; Garib, Z.; André, J.; Blackman, E.; Freeman, C.J.; Jorba, J.; et al. Outbreak of Poliomyelitis in Hispaniola Associated with Circulating Type 1 Vaccine-Derived Poliovirus. Science 2002, 296, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Flemister, M.R.; Brown, B.A.; Pallansch, M.A. Typing of human enteroviruses by partial sequencing of VP1. J. Clin. Microbiol. 1999, 37, 1288–1293. [Google Scholar] [CrossRef]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.; Sun, H.; Jeffries-Miles, S.; Gerloff, N.; Mandelbaum, M.; Pang, H.; Collins, N.; Burns, C.C.; Vega, E. Culture-Independent Detection of Poliovirus in Stool Samples by Direct RNA Extraction. Microbiol. Spectr. 2021, 9, e0066821. [Google Scholar] [CrossRef]
- Shaw, A.; Majumdar, M.; Troman, C.; O’Toole, Á.; Ansley, C.; Akello, J.; Bujaki, E.; Colquhoun, R.; Khurshida; Alammu; et al. Nested VP1 PCR and Nanopore Sequencing from Stool and ES Samples v1.2 V.3. 2022. Available online: https://www.protocols.io/view/nested-vp1-pcr-and-nanopore-sequencing-from-stool-81wgbpkmovpk/v3 (accessed on 30 August 2023).
- Kilpatrick, D.R.; Ching, K.; Iber, J.; Chen, Q.; Yang, S.-J.; De, L.; Williams, A.; Mandelbaum, M.; Sun, H.; Oberste, M.S.; et al. Identification of vaccine-derived polioviruses using dual-stage real-time RT-PCR. J. Virol. Methods 2013, 197, 25–28. [Google Scholar] [CrossRef]
- Kilpatrick, D.R.; Iber, J.C.; Chen, Q.; Ching, K.; Yang, S.-J.; De, L.; Mandelbaum, M.D.; Emery, B.; Campagnoli, R.; Burns, C.C.; et al. Poliovirus serotype-specific VP1 sequencing primers. J. Virol. Methods 2011, 174, 128–130. [Google Scholar] [CrossRef]
- Rotbart, H.A. Enzymatic RNA amplification of the enteroviruses. J. Clin. Microbiol. 1990, 28, 438–442. [Google Scholar] [CrossRef]
- Olive, D.M.; Al-Mufti, S.; Al-Mulla, W.; Khan, M.A.; Pasca, A.; Stanway, G.; Al-Nakib, W. Detection and differentiation of picornaviruses in clinical samples following genomic amplification. J. Gen. Virol. 1990, 71, 2141–2147. [Google Scholar] [CrossRef]
- National Institute of Infectious Diseases. The Laboratory Manual for Pathogen Detection of Hand, Foot and Mouth Disease (HFMD) (In Japanese). 2023. Available online: https://www.niid.go.jp/niid/images/lab-manual/HFMdis20230104.pdf (accessed on 30 August 2023).
- Poliovirus Sequencing Consortium. PIRANHA (Poliovirus Investigation Resource Automating Nanopore Haplotype Analysis). Available online: https://www.protocols.io/workspaces/poliovirus-sequencing-consortium/discussions/ddns-analysis-software (accessed on 30 August 2023).
- Dierssen, U.; Rehren, F.; Henke-Gendo, C.; Harste, G.; Heim, A. Rapid routine detection of enterovirus RNA in cerebrospinal fluid by a one-step real-time RT-PCR assay. J. Clin. Virol. 2008, 42, 58–64. [Google Scholar] [CrossRef]
- Sun, H.; Harrington, C.; Gerloff, N.; Mandelbaum, M.; Jeffries-Miles, S.; Apostol, L.N.G.; Valencia, M.A.-L.D.; Shaukat, S.; Angez, M.; Sharma, D.K.; et al. Validation of a redesigned pan-poliovirus assay and real-time PCR platforms for the global poliovirus laboratory network. PLoS ONE 2021, 16, e0255795. [Google Scholar] [CrossRef] [PubMed]
- Arita, M. Development of Poliovirus Extraction Method from Stool Extracts by Using Magnetic Nanoparticles Sensitized with Soluble Poliovirus Receptor. J. Clin. Microbiol. 2013, 51, 2717–2720. [Google Scholar] [CrossRef]
- Akello, J.O.; Bujaki, E.; Shaw, A.G.; Khurshid, A.; Arshad, Y.; Troman, C.; Majumdar, M.; O’Toole, Á.; Rambaut, A.; Alam, M.M.; et al. Comparison of Eleven RNA Extraction Methods for Poliovirus Direct Molecular Detection in Stool Samples. Microbiol. Spectr. 2023, 11, e0425222. [Google Scholar] [CrossRef] [PubMed]
- Honeywood, M.J.; Jeffries-Miles, S.; Wong, K.; Harrington, C.; Burns, C.C.; Oberste, M.S.; Bowen, M.D.; Vega, E. Use of guanidine thiocyanate-based nucleic acid extraction buffers to inactivate poliovirus in potentially infectious materials. J. Virol. Methods 2021, 297, 114262. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.G.; Mampuela, T.K.; Lofiko, E.L.; Pratt, C.; Troman, C.; Bujaki, E.; O’toole, Á.; Akello, J.O.; Aziza, A.A.; Lusamaki, E.K.; et al. Sensitive poliovirus detection using nested PCR and nanopore sequencing: A prospective validation study. Nat. Microbiol. 2023, 8, 1634–1640. [Google Scholar] [CrossRef] [PubMed]
- Klapsa, D.; Wilton, T.; Zealand, A.; Bujaki, E.; Saxentoff, E.; Troman, C.; Shaw, A.G.; Tedcastle, A.; Majumdar, M.; Mate, R.; et al. Sustained detection of type 2 poliovirus in London sewage between February and July, 2022, by enhanced environmental surveillance. Lancet 2022, 400, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Razafindratsimandresy, R.; Joffret, M.-L.; Andriamandimby, S.F.; Andriamamonjy, S.; Rabemanantsoa, S.; Richard, V.; Delpeyroux, F.; Heraud, J.-M.; Bessaud, M. Enterovirus detection in different regions of Madagascar reveals a higher abundance of enteroviruses of species C in areas where several outbreaks of vaccine-derived polioviruses occurred. BMC Infect. Dis. 2022, 22, 821. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Detection Method | Primer | Sequence (5′–3′) | Gene | Position * | Reference |
|---|---|---|---|---|---|
| EC RT-PCR | 5′NTR-529-552+ (5′NTR) | TGGCGGAACCGACTACTTTGGGTG | 5′NTR | 529–552 | [5] |
| Cre-4485-4460- (Cre) | TCAATACGGTGTTTGCTCTTGAACTG | 2C | 4460–4485 | ||
| Pan-PV-VP1 PCR | Y7 | GGGTTTGTGTCAGCCTGTAATGA | VP3 | 2399–2421 | [17] |
| Q8 | AAGAGGTCTCTRTTCCACAT | 2A | 3485–3504 | ||
| Type-specific VP1 PCR | 246S-S1 | CGAGATACCACACATATAGA | VP3 | 2441–2460 | [17] |
| 248S-S3 | CGAGACACCACTCACATTTC | VP3 | 2438–2457 | ||
| 252A-S1 | ATATGTGGTCAGATCCTTGGTG | VP1 | 3364–3385 | ||
| 254A-S3 | ATATGTGGTTAATCCTTTCTCA | VP1 | 3355–3376 | ||
| VP4–VP2 semi-nested RT-PCR | EVP2 | CCTCCGGCCCCTGAATGCGGCTAAT | 5′NTR | 444–468 | [18] |
| EVP4 | CTACTTTGGGTGTCCGTGTT | 5′NTR | 541–560 | [8] | |
| OL68-1 | GGTAAYTTCCACCACCANCC | VP2 | 1178–1197 | [19] |
| Samples | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |||||
| Number of copies of viral genome in 200 μL of stool suspension | 6.3 × 104 | 8.7 × 104 | 3.1 × 103 | 2.1 × 103 | 150 | ND | 160 | 1.1 × 103 | 18 | 94 | ||||
| RT-PCR kit | Method | Detection rate | Identification rate * | |||||||||||
| VI | Sabin 3 (L20B, 2nd passage) ** | Sabin 3 (L20B, 2nd passage) | Sabin 3 (L20B, day 4) | Sabin 3 (L20B, day 4) | Sabin 1 + Sabin 3 (L20B, day 4) | Sabin 1 + Sabin 3 (L20B, day 4) | Sabin 1 + Sabin 3 (L20B, day 4) | Sabin 1 + Sabin 3 (L20B, day 4) | Sabin 3 (RD, 2nd passage) | Sabin 3 (RD, 2nd passage) | ||||
| Kit A | EC RT-PCR + ITD | Ct value for Sabin 1 | 29.0 | 29.3 | 28.3 | 29.1 | 29.4 | 28.3 | 28.8 | 20.6 | 29.4 | 29.6 | 100% (10/10) | |
| Ct value for Sabin 3 | 10.6 | 11.3 | 13.5 | 13.3 | - | 30.3 | 17.5 | 11.9 | 32.9 | - | 80% (8/10) | |||
| Nanopore sequencing | Sabin 3 | Sabin 3 | Sabin 3 | Sabin 3 | - | - | Sabin 3 | Sabin 3 | - | - | 60% (6/10) | 40% (4/10) | ||
| Sanger sequencing | Sabin 1 + Sabin 3 | Sabin 3 | Sabin 1 + Sabin 3 | Sabin 3 | - | Sabin 3 | Sabin 3 | Sabin 1 + Sabin 3 | - | - | 70% (7/10) | 30% (3/10) | ||
| Kit B | EC RT-PCR + ITD | Ct value for Sabin 1 | - | - | - | - | 10.0 | - | - | - | - | - | 10% (1/10) | |
| Ct value for Sabin 3 | 8.6 | 6.0 | 6.3 | 6.4 | - | - | 9.8 | 10.1 | - | 10.5 | 70% (7/10) | |||
| Nanopore sequencing | Sabin 3 | Sabin 3 | Sabin 3 | Sabin 3 | Sabin 1 | - | Sabin 3 | Sabin 3 | - | Sabin 3 | 80% (8/10) | 50% (5/10) | ||
| Sanger sequencing | Sabin 3 | Sabin 3 | Sabin 3 | Sabin 3 | Sabin 1 | - | Sabin 3 | Sabin 3 | - | Sabin 3 | 80% (8/10) | 50% (5/10) | ||
| Sample | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||||
| Number of copies of viral genome in 200 μL of stool suspension | 6.3 × 104 | 8.7 × 104 | 3.1 × 103 | 2.1 × 103 | 150 | ND | 160 | 1.1 × 103 | 18 | 94 | |||
| Method | Detection rate | Identification rate * | |||||||||||
| VI | Sabin 3 | Sabin 3 | Sabin 3 | Sabin 3 | Sabin 1 + Sabin 3 | Sabin 1 + Sabin 3 | Sabin 1 + Sabin 3 | Sabin 1 + Sabin 3 | Sabin 3 | Sabin 3 | |||
| Sanger sequencing | Sabin 3 | Sabin 3 | Sabin 3 | Sabin 3 | Sabin 1 | Sabin 1 | Sabin 3 | Sabin 3 | Sabin 3 | Sabin 3 | 100% (10/10) | 60% (6/10) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueno, M.K.; Kitamura, K.; Nishimura, Y.; Arita, M. Evaluation of Direct Detection Protocols for Poliovirus from Stool Samples of Acute Flaccid Paralysis Patients. Viruses 2023, 15, 2113. https://doi.org/10.3390/v15102113
Ueno MK, Kitamura K, Nishimura Y, Arita M. Evaluation of Direct Detection Protocols for Poliovirus from Stool Samples of Acute Flaccid Paralysis Patients. Viruses. 2023; 15(10):2113. https://doi.org/10.3390/v15102113
Chicago/Turabian StyleUeno, Minami Kikuchi, Kouichi Kitamura, Yorihiro Nishimura, and Minetaro Arita. 2023. "Evaluation of Direct Detection Protocols for Poliovirus from Stool Samples of Acute Flaccid Paralysis Patients" Viruses 15, no. 10: 2113. https://doi.org/10.3390/v15102113
APA StyleUeno, M. K., Kitamura, K., Nishimura, Y., & Arita, M. (2023). Evaluation of Direct Detection Protocols for Poliovirus from Stool Samples of Acute Flaccid Paralysis Patients. Viruses, 15(10), 2113. https://doi.org/10.3390/v15102113