
RIOK3 and Its Alternatively Spliced Isoform Have Disparate Roles in the Innate Immune Response to Rift Valley Fever Virus (MP12) Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses, Infections and Cell Culture
2.2. Reagents
2.3. Plasmids, Oligos and Transfections
2.4. Luciferase Reporter Assays
2.5. RNA Extractions and Reverse Transcription
2.6. RT-qPCR
2.7. Gel Electrophoresis
2.8. Statistical Analysis
3. Results
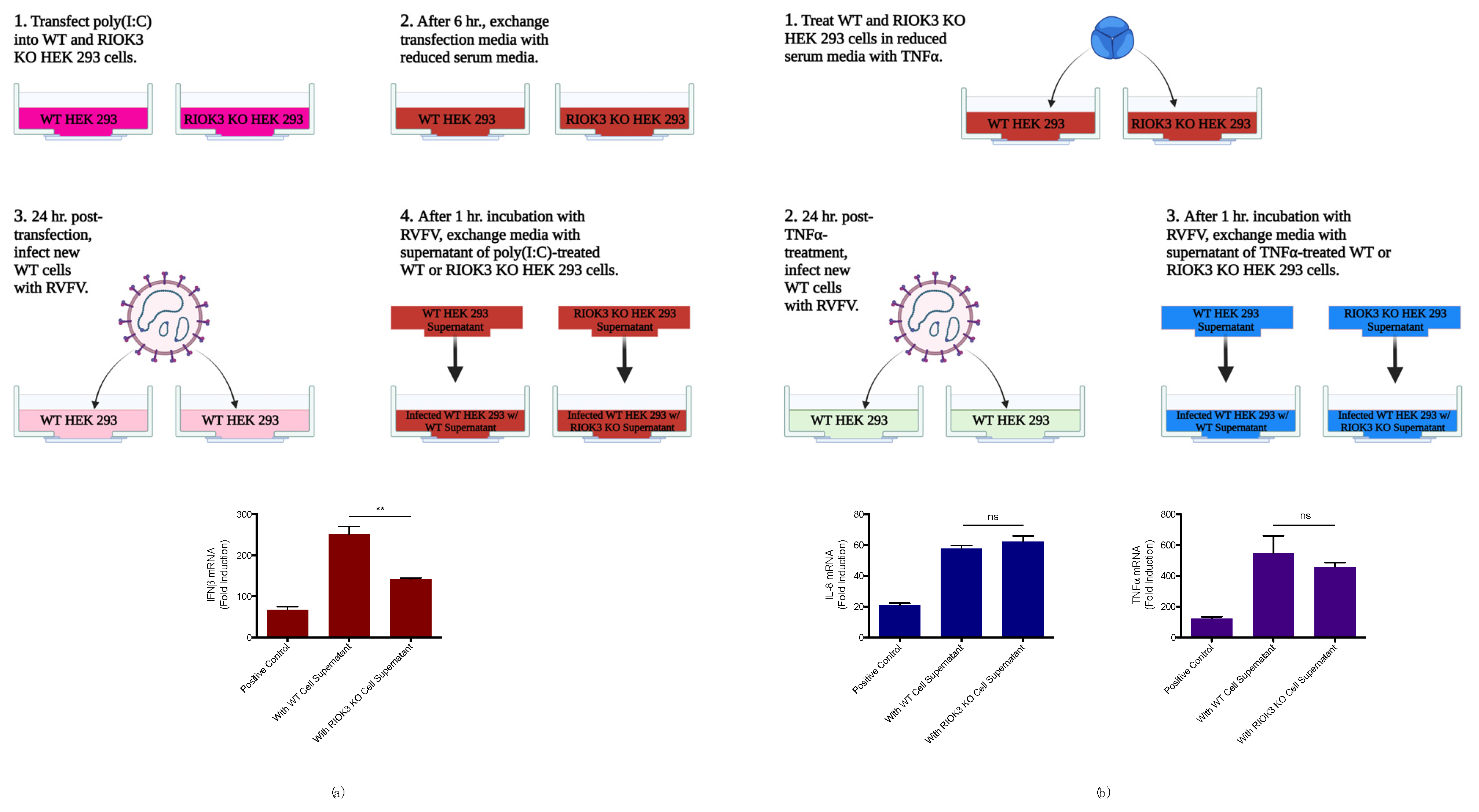
3.1. RIOK3 Has Distinct Roles in the Cellular IFN and NFκB Responses
3.2. RIOK3 Has a Role in Negatively Regulating the NFκB-Mediated Inflammatory Response during RVFV MP12 Infection
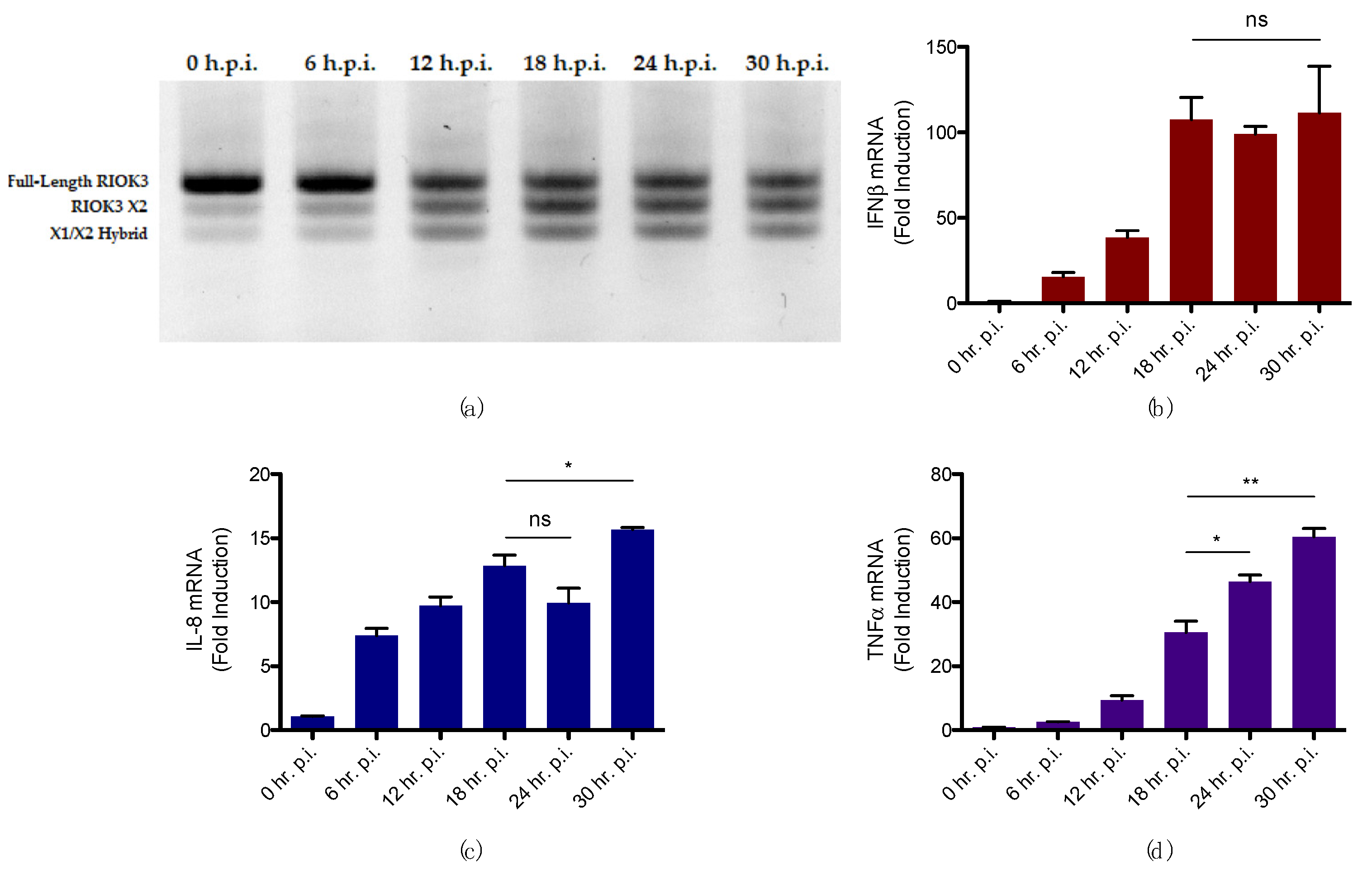
3.3. Alternative Splicing of RIOK3 during RVFV Infection Correlates with Increased Inflammatory Response and Decreased IFN Response
3.4. RIOK3 X2 Expression Augments TNFα-Induced Inflammatory Pathway Activation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, S. The Role of Ubiquitination in NF-kappaB Signaling during Virus Infection. Viruses 2021, 13, 145. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 2009, 284, 807–817. [Google Scholar] [CrossRef]
- Gao, D.; Yang, Y.K.; Wang, R.P.; Zhou, X.; Diao, F.C.; Li, M.D.; Zhai, Z.H.; Jiang, Z.F.; Chen, D.Y. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS ONE 2009, 4, e5760. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, M.; Chu, H.; Zhang, H.; Wu, H.; Song, G.; Wang, P.; Zhao, K.; Hou, J.; Wang, X.; et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 2017, 18, 214–224. [Google Scholar] [CrossRef]
- Zhou, Z.; Jia, X.; Xue, Q.; Dou, Z.; Ma, Y.; Zhao, Z.; Jiang, Z.; He, B.; Jin, Q.; Wang, J. TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proc. Natl. Acad. Sci. USA 2014, 111, E245–E254. [Google Scholar] [CrossRef]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef]
- Liu, H.M.; Loo, Y.M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The mitochondrial targeting chaperone 14-3-3epsilon regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Sun, L.; Liu, H.H.; Chen, X.; Seth, R.B.; Forman, J.; Chen, Z.J. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 2006, 24, 633–642. [Google Scholar] [CrossRef]
- de la Rica, R.; Borges, M.; Gonzalez-Freire, M. COVID-19: In the Eye of the Cytokine Storm. Front. Immunol. 2020, 11, 558898. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2017). Arch. Virol. 2017, 162, 2505–2538. [Google Scholar] [CrossRef]
- Collett, M.S.; Purchio, A.F.; Keegan, K.; Frazier, S.; Hays, W.; Anderson, D.K.; Parker, M.D.; Schmaljohn, C.; Schmidt, J.; Dalrymple, J.M. Complete nucleotide sequence of the M RNA segment of Rift Valley fever virus. Virology 1985, 144, 228–245. [Google Scholar] [CrossRef]
- Giorgi, C.; Accardi, L.; Nicoletti, L.; Gro, M.C.; Takehara, K.; Hilditch, C.; Morikawa, S.; Bishop, D.H. Sequences and coding strategies of the S RNAs of Toscana and Rift Valley fever viruses compared to those of Punta Toro, Sicilian Sandfly fever, and Uukuniemi viruses. Virology 1991, 180, 738–753. [Google Scholar] [CrossRef]
- Muller, R.; Poch, O.; Delarue, M.; Bishop, D.H.; Bouloy, M. Rift Valley fever virus L segment: Correction of the sequence and possible functional role of newly identified regions conserved in RNA-dependent polymerases. J. Gen. Virol. 1994, 75 Pt 6, 1345–1352. [Google Scholar] [CrossRef]
- NIAID. NIAID Emerging Infectious Diseases/Pathogens. Available online: https://www.niaid.nih.gov/research/emerging-infectious-diseases-pathogens (accessed on 15 July 2022).
- Vialat, P.; Muller, R.; Vu, T.H.; Prehaud, C.; Bouloy, M. Mapping of the mutations present in the genome of the Rift Valley fever virus attenuated MP12 strain and their putative role in attenuation. Virus Res. 1997, 52, 43–50. [Google Scholar] [CrossRef]
- Habjan, M.; Andersson, I.; Klingstrom, J.; Schumann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Muhlberger, E.; et al. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE 2008, 3, e2032. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M.; Weber, F. Bunyaviruses and the type I interferon system. Viruses 2009, 1, 1003–1021. [Google Scholar] [CrossRef]
- Havranek, K.E.; White, L.A.; Bisom, T.C.; Lanchy, J.M.; Lodmell, J.S. The Atypical Kinase RIOK3 Limits RVFV Propagation and Is Regulated by Alternative Splicing. Viruses 2021, 13, 367. [Google Scholar] [CrossRef]
- White, L.A.; Bisom, T.C.; Grimes, H.L.; Hayashi, M.; Lanchy, J.M.; Lodmell, J.S. Tra2beta-Dependent Regulation of RIO Kinase 3 Splicing During Rift Valley Fever Virus Infection Underscores the Links Between Alternative Splicing and Innate Antiviral Immunity. Front. Cell Infect. Microbiol. 2021, 11, 799024. [Google Scholar] [CrossRef] [PubMed]
- Havranek, K.E.; White, L.A.; Lanchy, J.M.; Lodmell, J.S. Transcriptome profiling in Rift Valley fever virus infected cells reveals modified transcriptional and alternative splicing programs. PLoS ONE 2019, 14, e0217497. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, S.M.; Fitzgerald, K.A.; Rosains, J.; Rowe, D.C.; Golenbock, D.T.; Maniatis, T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. USA 2004, 101, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ten Oever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell Biol. 1998, 18, 2986–2996. [Google Scholar] [CrossRef]
- Sato, M.; Tanaka, N.; Hata, N.; Oda, E.; Taniguchi, T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett. 1998, 425, 112–116. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [PubMed]
- Velazquez, L.; Fellous, M.; Stark, G.R.; Pellegrini, S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell 1992, 70, 313–322. [Google Scholar] [CrossRef]
- Müller, M.; Briscoe, J.; Laxton, C.; Guschin, D.; Ziemiecki, A.; Silvennoinen, O.; Harpur, A.G.; Barbieri, G.; Witthuhn, B.A.; Schindler, C.; et al. The protein tyrosine kinase JAK1 complements defects in interferon-alpha/beta and -gamma signal transduction. Nature 1993, 366, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.; Qureshi, S.A.; Kerr, I.M.; Darnell, J.E., Jr.; Stark, G.R. Role of STAT2 in the alpha interferon signaling pathway. Mol. Cell Biol. 1995, 15, 1312–1317. [Google Scholar] [CrossRef]
- Muller, M.; Laxton, C.; Briscoe, J.; Schindler, C.; Improta, T.; Darnell, J.E., Jr.; Stark, G.R.; Kerr, I.M. Complementation of a mutant cell line: Central role of the 91 kDa polypeptide of ISGF3 in the interferon-alpha and -gamma signal transduction pathways. EMBO J. 1993, 12, 4221–4228. [Google Scholar] [CrossRef]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef]
- Nan, Y.; Wu, C.; Zhang, Y.J. Interplay between Janus Kinase/Signal Transducer and Activator of Transcription Signaling Activated by Type I Interferons and Viral Antagonism. Front. Immunol. 2017, 8, 1758. [Google Scholar] [CrossRef]
- Feng, J.; De Jesus, P.D.; Su, V.; Han, S.; Gong, D.; Wu, N.C.; Tian, Y.; Li, X.; Wu, T.T.; Chanda, S.K.; et al. RIOK3 is an adaptor protein required for IRF3-mediated antiviral type I interferon production. J. Virol. 2014, 88, 7987–7997. [Google Scholar] [CrossRef]
- Takashima, K.; Oshiumi, H.; Takaki, H.; Matsumoto, M.; Seya, T. RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. 2015, 11, 192–200. [Google Scholar] [CrossRef]
- Shen, Y.; Tang, K.; Chen, D.; Hong, M.; Sun, F.; Wang, S.; Ke, Y.; Wu, T.; Sun, R.; Qian, J.; et al. Riok3 inhibits the antiviral immune response by facilitating TRIM40-mediated RIG-I and MDA5 degradation. Cell Rep. 2021, 35, 109272. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Baltimore, D. Activation of DNA-binding activity in an apparently cytoplasmic precursor of the NF-kappa B transcription factor. Cell 1988, 53, 211–217. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Baltimore, D. A 65-kappaD subunit of active NF-kappaB is required for inhibition of NF-kappaB by I kappaB. Genes Dev. 1989, 3, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Baltimore, D. I kappa B: A specific inhibitor of the NF-kappa B transcription factor. Science 1988, 242, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. NF-kappa B, KBF1, dorsal, and related matters. Cell 1990, 62, 841–843. [Google Scholar] [CrossRef]
- Rothwarf, D.M.; Zandi, E.; Natoli, G.; Karin, M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 1998, 395, 297–300. [Google Scholar] [CrossRef]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. IKK-1 and IKK-2: Cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef]
- Delhase, M.; Hayakawa, M.; Chen, Y.; Karin, M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science 1999, 284, 309–313. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 1997, 388, 548–554. [Google Scholar] [CrossRef]
- Zhao, T.; Yang, L.; Sun, Q.; Arguello, M.; Ballard, D.W.; Hiscott, J.; Lin, R. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat. Immunol. 2007, 8, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 1994, 78, 773–785. [Google Scholar] [CrossRef]
- Traenckner, E.B.; Wilk, S.; Baeuerle, P.A. A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of I kappa B-alpha that is still bound to NF-kappa B. EMBO J. 1994, 13, 5433–5441. [Google Scholar] [CrossRef]
- Brown, K.; Gerstberger, S.; Carlson, L.; Franzoso, G.; Siebenlist, U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 1995, 267, 1485–1488. [Google Scholar] [CrossRef] [PubMed]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef]
- Osborn, L.; Kunkel, S.; Nabel, G.J. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc. Natl. Acad. Sci. USA 1989, 86, 2336–2340. [Google Scholar] [CrossRef]
- Duh, E.J.; Maury, W.J.; Folks, T.M.; Fauci, A.S.; Rabson, A.B. Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc. Natl. Acad. Sci. USA 1989, 86, 5974–5978. [Google Scholar] [CrossRef] [PubMed]
- Lowenthal, J.W.; Ballard, D.W.; Bohnlein, E.; Greene, W.C. Tumor necrosis factor alpha induces proteins that bind specifically to kappa B-like enhancer elements and regulate interleukin 2 receptor alpha-chain gene expression in primary human T lymphocytes. Proc. Natl. Acad. Sci. USA 1989, 86, 2331–2335. [Google Scholar] [CrossRef]
- Shakhov, A.N.; Collart, M.A.; Vassalli, P.; Nedospasov, S.A.; Jongeneel, C.V. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J. Exp. Med. 1990, 171, 35–47. [Google Scholar] [CrossRef]
- Shan, J.; Wang, P.; Zhou, J.; Wu, D.; Shi, H.; Huo, K. RIOK3 interacts with caspase-10 and negatively regulates the NF-kappaB signaling pathway. Mol. Cell Biochem. 2009, 332, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Fenner, B.J.; Scannell, M.; Prehn, J.H. Identification of polyubiquitin binding proteins involved in NF-kappaB signaling using protein arrays. Biochim. Biophys. Acta 2009, 1794, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, J.; Wicht, O.; Wolanski, J.C.; Baur, N.; Bastian, S.; Haas, D.A.; Matula, P.; Knapp, B.; Meyniel-Schicklin, L.; Wang, C.; et al. Phosphorylation-Dependent Feedback Inhibition of RIG-I by DAPK1 Identified by Kinome-wide siRNA Screening. Mol. Cell 2017, 65, 403–415.e8. [Google Scholar] [CrossRef]
- Smith, M.R.; Schirtzinger, E.E.; Wilson, W.C.; Davis, A.S. Rift Valley Fever Virus: Propagation, Quantification, and Storage. Curr. Protoc. Microbiol. 2019, 55, e92. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-kappaB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef]
- De Waele, J.; Verhezen, T.; van der Heijden, S.; Berneman, Z.N.; Peeters, M.; Lardon, F.; Wouters, A.; Smits, E. A systematic review on poly(I:C) and poly-ICLC in glioblastoma: Adjuvants coordinating the unlocking of immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 213. [Google Scholar] [CrossRef]
- Kunsch, C.; Rosen, C.A. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol. Cell Biol. 1993, 13, 6137–6146. [Google Scholar]
- Mazumder, B.; Li, X.; Barik, S. Translation control: A multifaceted regulator of inflammatory response. J. Immunol. 2010, 184, 3311–3319. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Cerikan, B.; Cortese, M.; Frankish, J.; Lee, J.Y.; Plociennikowska, A.; Heigwer, F.; Prasad, V.; Joecks, S.; Burkart, S.S.; et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-kappaB. Commun. Biol. 2022, 5, 45. [Google Scholar] [CrossRef]
- Nilsson-Payant, B.E.; Uhl, S.; Grimont, A.; Doane, A.S.; Cohen, P.; Patel, R.S.; Higgins, C.A.; Acklin, J.A.; Bram, Y.; Chandar, V.; et al. The NF-kappaB Transcriptional Footprint Is Essential for SARS-CoV-2 Replication. J. Virol. 2021, 95, e0125721. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Liu, T.; Ko, C.J.; Zhang, L.; Joo, D.; Xie, X.; Zhu, L.; Li, Y.; Cheng, X.; Sun, S.C. Myeloid cell TBK1 restricts inflammatory responses. Proc. Natl. Acad. Sci. USA 2022, 119, e2107742119. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Lu, M.; Tian, B.; Li, K.; Garofalo, R.P.; Prusak, D.; Wood, T.G.; Brasier, A.R. Expression of an IKKgamma splice variant determines IRF3 and canonical NF-kappaB pathway utilization in ssRNA virus infection. PLoS ONE 2009, 4, e8079. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisom, T.C.; White, L.A.; Lanchy, J.-M.; Lodmell, J.S. RIOK3 and Its Alternatively Spliced Isoform Have Disparate Roles in the Innate Immune Response to Rift Valley Fever Virus (MP12) Infection. Viruses 2022, 14, 2064. https://doi.org/10.3390/v14092064
Bisom TC, White LA, Lanchy J-M, Lodmell JS. RIOK3 and Its Alternatively Spliced Isoform Have Disparate Roles in the Innate Immune Response to Rift Valley Fever Virus (MP12) Infection. Viruses. 2022; 14(9):2064. https://doi.org/10.3390/v14092064
Chicago/Turabian StyleBisom, Thomas C., Luke A. White, Jean-Marc Lanchy, and J. Stephen Lodmell. 2022. "RIOK3 and Its Alternatively Spliced Isoform Have Disparate Roles in the Innate Immune Response to Rift Valley Fever Virus (MP12) Infection" Viruses 14, no. 9: 2064. https://doi.org/10.3390/v14092064
APA StyleBisom, T. C., White, L. A., Lanchy, J.-M., & Lodmell, J. S. (2022). RIOK3 and Its Alternatively Spliced Isoform Have Disparate Roles in the Innate Immune Response to Rift Valley Fever Virus (MP12) Infection. Viruses, 14(9), 2064. https://doi.org/10.3390/v14092064