
Antiretroviral Drug Discovery Targeting the HIV-1 Nef Virulence Factor
 , , ,
, , ,
Abstract
1. Introduction
2. HIV-1 Nef as a Rational Target for Antiretroviral Drug Development
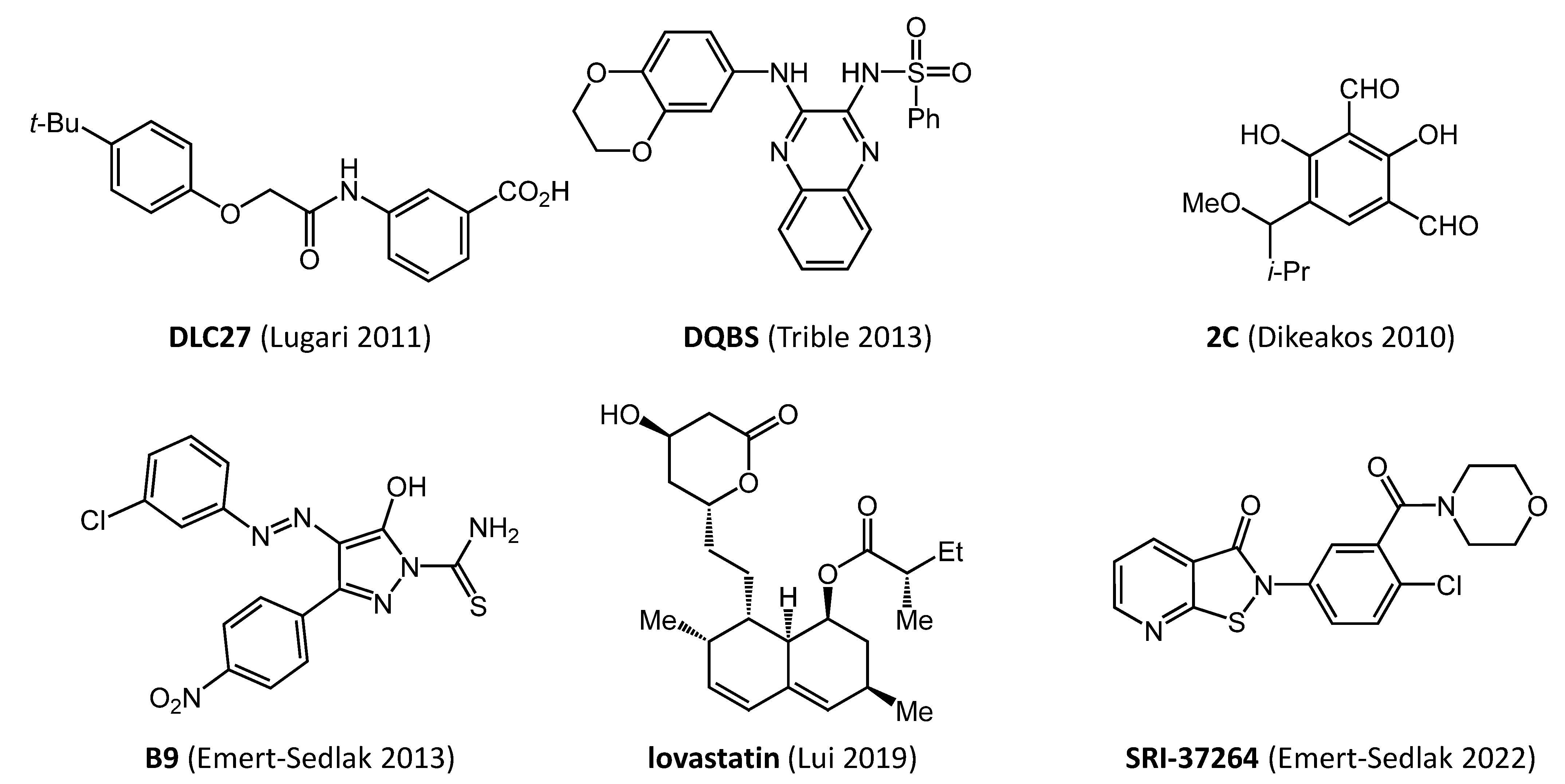
3. Early Efforts in Nef Drug Discovery

{kind=link}
{kind=link}
| Compound | Direct Nef Binding | Inhibits HIV-1 Infectivity | Inhibits HIV-1 Replication | Restores Cell-Surface MHC-I | Triggers Anti-HIV CTL Response | Ref. |
|---|---|---|---|---|---|---|
| DLC-27 | yes | nd | nd | yes | nd | [27] |
| 2C | yes | yes | nd | yes | nd | [31] |
| lovastatin | yes | yes | nd | yes | yes | [32] |
| concanamycin A | no | nd | nd | yes | yes | [33] |
| DFP analogs | no | yes | yes | nd | nd | [34] |
| B9 | yes | yes | yes | yes | yes | [35,36] |
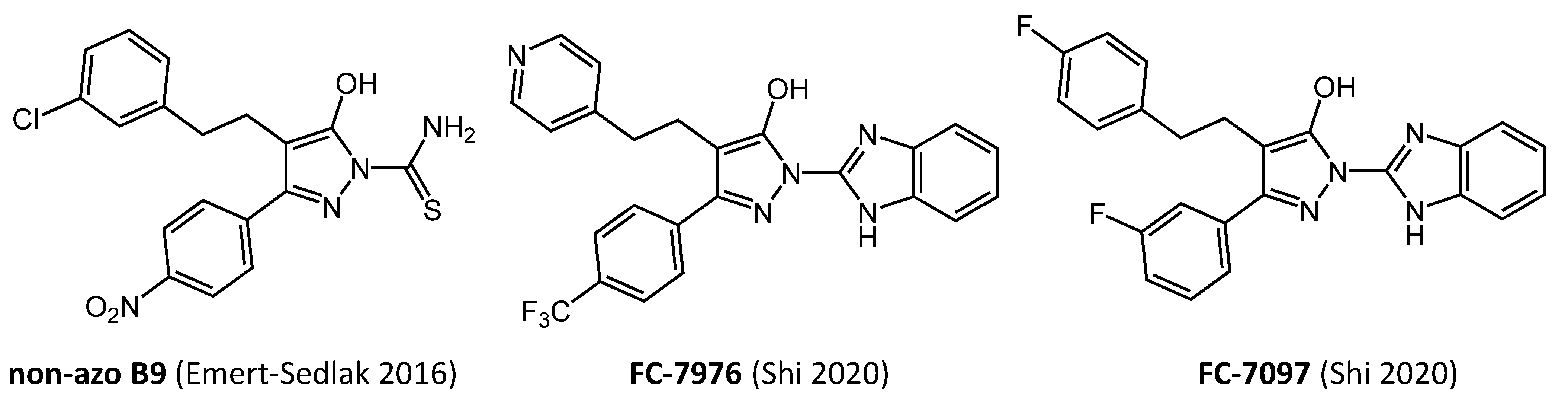
| non-azo B9 | yes | yes | yes | yes | yes | [35,36,37] |
| SRI-37264 | yes | yes | yes | yes | nd | [38] |
| FC-7976 | yes | yes | yes | yes | nd | [39] |
| DQBS | yes | yes | yes | yes | nd | [40] |
4. Natural Products and Repurposed Drugs Inhibit Nef-Dependent Downregulation of MHC-I
5. Coupling Nef to Src-Family Kinase Activation Enables Discovery of Direct Nef Antagonists
6. Hydroxypyrazole Nef inhibitor Analogs Display Potent Antiretroviral Activity in Primary Cells and Restore Cell-Surface MHC-I Expression
7. Mechanism of Action of Hydroxypyrazole Nef Inhibitors—Clues from Structural Biology
8. Alternative Approaches to Nef Drug Discovery
9. Summary and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dionne, B. Key Principles of Antiretroviral Pharmacology. Infect. Dis. Clin. N. Am. 2019, 33, 787–805. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; De Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef] [PubMed]
- McBrien, J.B.; Mavigner, M.; Franchitti, L.; Smith, S.A.; White, E.; Tharp, G.K.; Walum, H.; Busman-Sahay, K.; Aguilera-Sandoval, C.R.; Thayer, W.O.; et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8(+) cells. Nature 2020, 578, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-kappaB signalling in vivo. Nature 2020, 578, 160–165. [Google Scholar] [CrossRef]
- Geyer, M.; Munte, C.E.; Schorr, J.; Kellner, R.; Kalbitzer, H.R. Structure of the anchor-domain of myristoylated and non-myristoylated HIV-1 Nef protein. J. Mol. Biol. 1999, 289, 123–138. [Google Scholar] [CrossRef]
- Gerlach, H.; Laumann, V.; Martens, S.; Becker, C.F.; Goody, R.S.; Geyer, M. HIV-1 Nef membrane association depends on charge, curvature, composition and sequence. Nat. Chem. Biol. 2010, 6, 46–53. [Google Scholar] [CrossRef]
- Akgun, B.; Satija, S.; Nanda, H.; Pirrone, G.F.; Shi, X.; Engen, J.R.; Kent, M.S. Conformational transition of membrane-associated terminally acylated HIV-1 Nef. Structure 2013, 21, 1822–1833. [Google Scholar] [CrossRef]
- Kent, M.S.; Murton, J.K.; Sasaki, D.Y.; Satija, S.; Akgun, B.; Nanda, H.; Curtis, J.E.; Majewski, J.; Morgan, C.R.; Engen, J.R. Neutron reflectometry study of the conformation of HIV Nef bound to lipid membranes. Biophys. J. 2010, 99, 1940–1948. [Google Scholar] [CrossRef]
- Pawlak, E.N.; Dikeakos, J.D. HIV-1 Nef: A master manipulator of the membrane trafficking machinery mediating immune evasion. Biochim. Biophys. Acta 2015, 1850, 733–741. [Google Scholar] [CrossRef]
- Doria, M. Role of the CD4 down-modulation activity of Nef in HIV-1 infectivity. Curr. HIV Res. 2011, 9, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Bregnard, C.; Zamborlini, A.; Leduc, M.; Chafey, P.; Camoin, L.; Saib, A.; Benichou, S.; Danos, O.; Basmaciogullari, S. Comparative proteomic analysis of HIV-1 particles reveals a role for Ezrin and EHD4 in the Nef-dependent increase of virus infectivity. J. Virol. 2013, 87, 3729–3740. [Google Scholar] [CrossRef]
- Fackler, O.T.; Luo, W.; Geyer, M.; Alberts, A.S.; Peterlin, B.M. Activation of Vav by Nef induces cytoskeletal rearrangements and downstream effector functions. Mol. Cell 1999, 3, 729–739. [Google Scholar] [CrossRef]
- Staudt, R.P.; Alvarado, J.J.; Emert-Sedlak, L.A.; Shi, H.; Shu, S.T.; Wales, T.E.; Engen, J.R.; Smithgall, T.E. Structure, function, and inhibitor targeting of HIV-1 Nef-effector kinase complexes. J. Biol. Chem. 2020, 295, 15158–15171. [Google Scholar] [CrossRef]
- Kestler, H.; Ringler, D.J.; Mori, K.; Panicali, D.L.; Sehgal, P.K.; Daniel, M.D.; Desrosiers, R.C. Importance of the nef gene for maintenance of high viral loads and for development of AIDS. Cell 1991, 65, 651–662. [Google Scholar] [CrossRef]
- Kirchhoff, F.; Greenough, T.C.; Brettler, D.B.; Sullivan, J.L.; Desrosiers, R.C. Absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 1995, 332, 228–232. [Google Scholar] [CrossRef]
- Deacon, N.J.; Tsykin, A.; Solomon, A.; Smith, K.; Ludford-Menting, M.; Hooker, D.J.; McPhee, D.A.; Greenway, A.L.; Ellett, A.; Chatfield, C.; et al. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 1995, 270, 988–991. [Google Scholar] [CrossRef]
- Zou, W.; Denton, P.W.; Watkins, R.L.; Krisko, J.F.; Nochi, T.; Foster, J.L.; Garcia, J.V. Nef functions in BLT mice to enhance HIV-1 replication and deplete CD4+CD8+ thymocytes. Retrovirology 2012, 9, 44. [Google Scholar] [CrossRef]
- Watkins, R.L.; Foster, J.L.; Garcia, J.V. In vivo analysis of Nef’s role in HIV-1 replication, systemic T cell activation and CD4(+) T cell loss. Retrovirology 2015, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.; Fackler, O.T.; Peterlin, B.M. Structure—Function relationships in HIV-1 Nef. EMBO Rep. 2001, 2, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Saksela, K. Interactions of the HIV/SIV pathogenicity factor Nef with SH3 domain-containing host cell proteins. Curr. HIV Res. 2011, 9, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Saksela, K.; Cheng, G.; Baltimore, D. Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. EMBO J. 1995, 14, 484–491. [Google Scholar] [CrossRef]
- Lee, C.-H.; Saksela, K.; Mirza, U.A.; Chait, B.T.; Kuriyan, J. Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell 1996, 85, 931–942. [Google Scholar] [CrossRef]
- Zhao, Z.; Fagerlund, R.; Tossavainen, H.; Hopfensperger, K.; Lotke, R.; Srinivasachar Badarinarayan, S.; Kirchhoff, F.; Permi, P.; Sato, K.; Sauter, D.; et al. Evolutionary plasticity of SH3 domain binding by Nef proteins of the HIV-1/SIVcpz lentiviral lineage. PLoS Pathog. 2021, 17, e1009728. [Google Scholar] [CrossRef]
- Betzi, S.; Restouin, A.; Opi, S.; Arold, S.T.; Parrot, I.; Guerlesquin, F.; Morelli, X.; Collette, Y. Protein protein interaction inhibition (2P2I) combining high throughput and virtual screening: Application to the HIV-1 Nef protein. Proc. Natl. Acad. Sci. USA 2007, 104, 19256–19261. [Google Scholar] [CrossRef]
- Jia, X.; Singh, R.; Homann, S.; Yang, H.; Guatelli, J.; Xiong, Y. Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nat. Struct. Mol. Biol. 2012, 19, 701–706. [Google Scholar] [CrossRef]
- Lugari, A.; Breuer, S.; Coursindel, T.; Opi, S.; Restouin, A.; Shi, X.; Nazabal, A.; Torbett, B.E.; Martinez, J.; Collette, Y.; et al. A specific protein disorder catalyzer of HIV-1 Nef. Bioorg. Med. Chem. 2011, 19, 7401–7406. [Google Scholar] [CrossRef]
- Lurie, A.; Fink, C.; Gosselin, G.; Dekaban, G.A.; Dikeakos, J.D. Inhibitors of HIV-1 Nef: Applications and developments for a practical cure. Virologie 2022, 26, 17–33. [Google Scholar] [CrossRef]
- Dikeakos, J.D.; Atkins, K.M.; Thomas, L.; Emert-Sedlak, L.; Byeon, I.J.; Jung, J.; Ahn, J.; Wortman, M.D.; Kukull, B.; Saito, M.; et al. Small molecule inhibition of HIV-1-induced MHC-I down-regulation identifies a temporally regulated switch in Nef action. Mol. Biol. Cell 2010, 21, 3279–3292. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, X.; Zhang, W.; Wu, L.; Jing, S.; Liu, W.; Xia, B.; Zou, F.; Lu, L.; Ma, X.; et al. Lovastatin Inhibits HIV-1-Induced MHC-I Downregulation by Targeting Nef-AP-1 Complex Formation: A New Strategy to Boost Immune Eradication of HIV-1 Infected Cells. Front. Immunol. 2019, 10, 2151. [Google Scholar] [CrossRef] [PubMed]
- Painter, M.M.; Zimmerman, G.E.; Merlino, M.S.; Robertson, A.W.; Terry, V.H.; Ren, X.; McLeod, M.R.; Gomez-Rodriguez, L.; Garcia, K.A.; Leonard, J.A.; et al. Concanamycin A counteracts HIV-1 Nef to enhance immune clearance of infected primary cells by cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 23835–23846. [Google Scholar] [CrossRef]
- Emert-Sedlak, L.; Kodama, T.; Lerner, E.C.; Dai, W.; Foster, C.; Day, B.W.; Lazo, J.S.; Smithgall, T.E. Chemical library screens targeting an HIV-1 accessory factor/host cell kinase complex identify novel antiretroviral compounds. ACS Chem. Biol. 2009, 4, 939–947. [Google Scholar] [CrossRef]
- Emert-Sedlak, L.A.; Narute, P.; Shu, S.T.; Poe, J.A.; Shi, H.; Yanamala, N.; Alvarado, J.J.; Lazo, J.S.; Yeh, J.I.; Johnston, P.A.; et al. Effector Kinase Coupling Enables High-Throughput Screens for Direct HIV-1 Nef Antagonists with Antiretroviral Activity. Chem. Biol. 2013, 20, 82–91. [Google Scholar] [CrossRef]
- Mujib, S.; Saiyed, A.; Fadel, S.; Bozorgzad, A.; Aidarus, N.; Yue, F.Y.; Benko, E.; Kovacs, C.; Emert-Sedlak, L.; Smithgall, T.E.; et al. Pharmacologic HIV-1 Nef Blockade Enhances the Recognition and Elimination of Latently HIV-1 Infected CD4 T cells by Autologous CD8 T cells. J. Clin. Investig. Insight 2017, 2, e93684. [Google Scholar]
- Emert-Sedlak, L.A.; Loughran, H.M.; Shi, H.; Kulp, J.L., III; Shu, S.T.; Zhao, J.; Day, B.W.; Wrobel, J.E.; Reitz, A.B.; Smithgall, T.E. Synthesis and evaluation of orally active small molecule HIV-1 Nef antagonists. Bioorg. Med. Chem. Lett. 2016, 26, 1480–1484. [Google Scholar] [CrossRef]
- Emert-Sedlak, L.A.; Moukha-Chafiq, O.; Shi, H.; Du, S.; Alvarado, J.J.; Pathak, V.; Tanner, S.G.; Hunter, R.N.; Nebane, M.; Chen, L.; et al. Inhibitors of HIV-1 Nef-Mediated Activation of the Myeloid Src-Family Kinase Hck Block HIV-1 Replication in Macrophages and Disrupt MHC-I Downregulation. ACS Infect. Dis. 2022, 8, 91–105. [Google Scholar] [CrossRef]
- Shi, H.; Tice, C.M.; Emert-Sedlak, L.; Chen, L.; Li, W.F.; Carlsen, M.; Wrobel, J.E.; Reitz, A.B.; Smithgall, T.E. Tight-Binding Hydroxypyrazole HIV-1 Nef Inhibitors Suppress Viral Replication in Donor Mononuclear Cells and Reverse Nef-Mediated MHC-I Downregulation. ACS Infect. Dis. 2020, 6, 10. [Google Scholar] [CrossRef]
- Trible, R.P.; Narute, P.; Emert-Sedlak, L.A.; Alvarado, J.J.; Atkins, K.; Thomas, L.; Kodama, T.; Yanamala, N.; Korotchenko, V.; Day, B.W.; et al. Discovery of a diaminoquinoxaline benzenesulfonamide antagonist of HIV-1 Nef function using a yeast-based phenotypic screen. Retrovirology 2013, 10, 135. [Google Scholar] [CrossRef]
- Duette, G.; Hiener, B.; Morgan, H.; Mazur, F.G.; Mathivanan, V.; Horsburgh, B.A.; Fisher, K.; Tong, O.; Lee, E.; Ahn, H.; et al. The HIV-1 proviral landscape reveals that Nef contributes to HIV-1 persistence in effector memory CD4+ T cells. J. Clin. Investig. 2022, 132, e154422. [Google Scholar] [CrossRef] [PubMed]
- Omondi, F.H.; Chandrarathna, S.; Mujib, S.; Brumme, C.J.; Jin, S.W.; Sudderuddin, H.; Miller, R.L.; Rahimi, A.; Laeyendecker, O.; Bonner, P.; et al. HIV Subtype and Nef-Mediated Immune Evasion Function Correlate with Viral Reservoir Size in Early-Treated Individuals. J. Virol. 2019, 93, e01832. [Google Scholar] [CrossRef] [PubMed]
- Atkins, K.M.; Thomas, L.; Youker, R.T.; Harriff, M.J.; Pissani, F.; You, H.; Thomas, G. HIV-1 Nef binds PACS-2 to assemble a multikinase cascade that triggers major histocompatibility complex class I (MHC-I) down-regulation: Analysis using short interfering RNA and knock-out mice. J. Biol. Chem. 2008, 283, 11772–11784. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Thomas, L.; Ruby, C.E.; Atkins, K.M.; Morris, N.P.; Knight, Z.A.; Scholz, I.; Barklis, E.; Weinberg, A.D.; Shokat, K.M.; et al. HIV-1 Nef assembles a Src family kinase-ZAP-70/Syk-PI3K cascade to downregulate cell-surface MHC-I. Cell Host Microbe 2007, 1, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.R.; Roeth, J.F.; Williams, M.; Filzen, T.M.; Fleis, R.I.; Collins, K.L. HIV-1 Nef disrupts antigen presentation early in the secretory pathway. J. Biol. Chem. 2005, 280, 12840–12848. [Google Scholar] [CrossRef]
- Pereira, E.A.; daSilva, L.L. HIV-1 Nef: Taking Control of Protein Trafficking. Traffic 2016, 17, 976–996. [Google Scholar] [CrossRef]
- Oneyama, C.; Nakano, H.; Sharma, S.V. UCS15A, a novel small molecule, SH3 domain-mediated protein-protein interaction blocking drug. Oncogene 2002, 21, 2037–2050. [Google Scholar] [CrossRef]
- Oneyama, C.; Agatsuma, T.; Kanda, Y.; Nakano, H.; Sharma, S.V.; Nakano, S.; Narazaki, F.; Tatsuta, K. Synthetic inhibitors of proline-rich ligand-mediated protein-protein interaction: Potent analogs of UCS15A. Chem. Biol. 2003, 10, 443–451. [Google Scholar] [CrossRef]
- Trible, R.P.; Emert-Sedlak, L.; Smithgall, T.E. HIV-1 Nef selectively activates SRC family kinases HCK, LYN, and c-SRC through direct SH3 domain interaction. J. Biol. Chem. 2006, 281, 27029–27038. [Google Scholar] [CrossRef]
- Chutiwitoonchai, N.; Hiyoshi, M.; Mwimanzi, P.; Ueno, T.; Adachi, A.; Ode, H.; Sato, H.; Fackler, O.T.; Okada, S.; Suzu, S. The identification of a small molecule compound that reduces HIV-1 Nef-mediated viral infectivity enhancement. PLoS ONE 2011, 6, e27696. [Google Scholar] [CrossRef]
- Montoya, C.J.; Higuita, E.A.; Estrada, S.; Gutierrez, F.J.; Amariles, P.; Giraldo, N.A.; Jimenez, M.M.; Velasquez, C.P.; Leon, A.L.; Rugeles, M.T.; et al. Randomized clinical trial of lovastatin in HIV-infected, HAART naive patients (NCT00721305). J. Infect. 2012, 65, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Guiet, R.; Poincloux, R.; Castandet, J.; Marois, L.; Labrousse, A.; Le Cabec, V.; Maridonneau-Parini, I. Hematopoietic cell kinase (Hck) isoforms and phagocyte duties—From signaling and actin reorganization to migration and phagocytosis. Eur. J. Cell Biol. 2008, 87, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Komuro, I.; Yokota, Y.; Yasuda, S.; Iwamoto, A.; Kagawa, K.S. CSF-induced and HIV-1-mediated distinct regulation of Hck and C/EBPbeta represent a heterogeneous susceptibility of monocyte-derived macrophages to M-tropic HIV-1 infection. J. Exp. Med. 2003, 198, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Engen, J.R.; Wales, T.E.; Hochrein, J.M.; Meyn, M.A., III; Banu, O.S.; Bahar, I.; Smithgall, T.E. Structure and dynamic regulation of Src-family kinases. Cell Mol. Life Sci 2008, 65, 3058–3073. [Google Scholar] [CrossRef]
- Boggon, T.J.; Eck, M.J. Structure and regulation of Src family kinases. Oncogene 2004, 23, 7918–7927. [Google Scholar] [CrossRef]
- Moarefi, I.; LaFevre-Bernt, M.; Sicheri, F.; Huse, M.; Lee, C.-H.; Kuriyan, J.; Miller, W.T. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature 1997, 385, 650–653. [Google Scholar] [CrossRef]
- Briggs, S.D.; Sharkey, M.; Stevenson, M.; Smithgall, T.E. SH3-mediated Hck tyrosine kinase activation and fibroblast transformation by the Nef protein of HIV-1. J. Biol. Chem. 1997, 272, 17899–17902. [Google Scholar] [CrossRef]
- Wales, T.E.; Hochrein, J.M.; Morgan, C.R.; Emert-Sedlak, L.A.; Smithgall, T.E.; Engen, J.R. Subtle Dynamic Changes Accompany Hck Activation by HIV-1 Nef and are Reversed by an Antiretroviral Kinase Inhibitor. Biochemistry 2015, 54, 6382–6391. [Google Scholar] [CrossRef]
- Lerner, E.C.; Smithgall, T.E. SH3-dependent stimulation of Src-family kinase autophosphorylation without tail release from the SH2 domain in vivo. Nat. Struct. Biol. 2002, 9, 365–369. [Google Scholar] [CrossRef]
- Narute, P.S.; Smithgall, T.E. Nef alleles from all major HIV-1 clades activate Src-family kinases and enhance HIV-1 replication in an inhibitor-sensitive manner. PLoS ONE 2012, 7, e32561. [Google Scholar] [CrossRef]
- Anmole, G.; Kuang, X.T.; Toyoda, M.; Martin, E.; Shahid, A.; Le, A.Q.; Markle, T.; Baraki, B.; Jones, R.B.; Ostrowski, M.A.; et al. A robust and scalable TCR-based reporter cell assay to measure HIV-1 Nef-mediated T cell immune evasion. J. Immunol. Methods 2015, 426, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, J.J.; Tarafdar, S.; Yeh, J.I.; Smithgall, T.E. Interaction with the Src homology (SH3-SH2) region of the Src-family kinase Hck structures the HIV-1 Nef dimer for kinase activation and effector recruitment. J. Biol. Chem. 2014, 289, 28539–28553. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Kaake, R.M.; Echeverria, I.; Suarez, M.; Karimian Shamsabadi, M.; Stoneham, C.; Ramirez, P.W.; Kress, J.; Singh, R.; Sali, A.; et al. Structural basis of CD4 downregulation by HIV-1 Nef. Nat. Struct. Mol. Biol. 2020, 27, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Moroco, J.A.; Alvarado, J.J.; Staudt, R.P.; Wales, T.E.; Smithgall, T.E.; Engen, J.R. Remodeling of HIV-1 Nef Structure by Src-Family Kinase Binding. J. Mol. Biol. 2018, 430, 310–321. [Google Scholar] [CrossRef]
- Poe, J.A.; Vollmer, L.; Vogt, A.; Smithgall, T.E. Development and Validation of a High-Content Bimolecular Fluorescence Complementation Assay for Small-Molecule Inhibitors of HIV-1 Nef Dimerization. J. Biomol. Screen 2014, 19, 556–565. [Google Scholar] [CrossRef]
- Romei, M.G.; Boxer, S.G. Split Green Fluorescent Proteins: Scope, Limitations, and Outlook. Annu. Rev. Biophys. 2019, 48, 19–44. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emert-Sedlak, L.A.; Shi, H.; Tice, C.M.; Chen, L.; Alvarado, J.J.; Shu, S.T.; Du, S.; Thomas, C.E.; Wrobel, J.E.; Reitz, A.B.; et al. Antiretroviral Drug Discovery Targeting the HIV-1 Nef Virulence Factor. Viruses 2022, 14, 2025. https://doi.org/10.3390/v14092025
Emert-Sedlak LA, Shi H, Tice CM, Chen L, Alvarado JJ, Shu ST, Du S, Thomas CE, Wrobel JE, Reitz AB, et al. Antiretroviral Drug Discovery Targeting the HIV-1 Nef Virulence Factor. Viruses. 2022; 14(9):2025. https://doi.org/10.3390/v14092025
Chicago/Turabian StyleEmert-Sedlak, Lori A., Haibin Shi, Colin M. Tice, Li Chen, John J. Alvarado, Sherry T. Shu, Shoucheng Du, Catherine E. Thomas, Jay E. Wrobel, Allen B. Reitz, and et al. 2022. "Antiretroviral Drug Discovery Targeting the HIV-1 Nef Virulence Factor" Viruses 14, no. 9: 2025. https://doi.org/10.3390/v14092025
APA StyleEmert-Sedlak, L. A., Shi, H., Tice, C. M., Chen, L., Alvarado, J. J., Shu, S. T., Du, S., Thomas, C. E., Wrobel, J. E., Reitz, A. B., & Smithgall, T. E. (2022). Antiretroviral Drug Discovery Targeting the HIV-1 Nef Virulence Factor. Viruses, 14(9), 2025. https://doi.org/10.3390/v14092025