
Computational and Experimental Approaches to Study the RNA Secondary Structures of RNA Viruses




Abstract
:1. Introduction
2. Existing Computational and Experimental Approaches to Mapping Viral RNA Structures
3. Computational Approaches to Studying RNA Virus Genome Structures
4. Experimental Approaches to Studying RNA Virus Genome Structures
5. Combining Experimental and Computational Approaches to Identify Functional RNA Structures
6. The Structurome of RNA Viruses Involved in Pandemics
7. Discussion

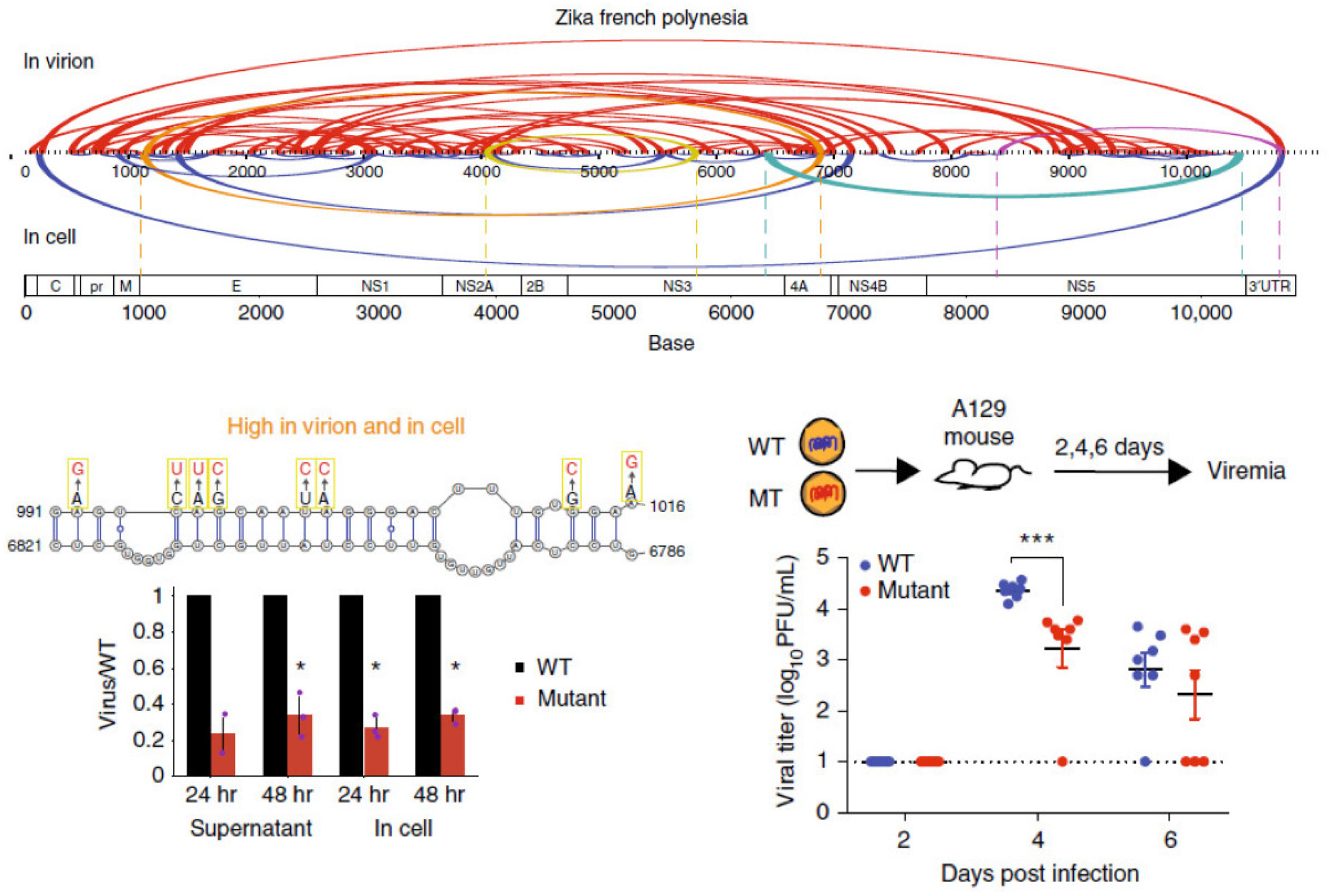
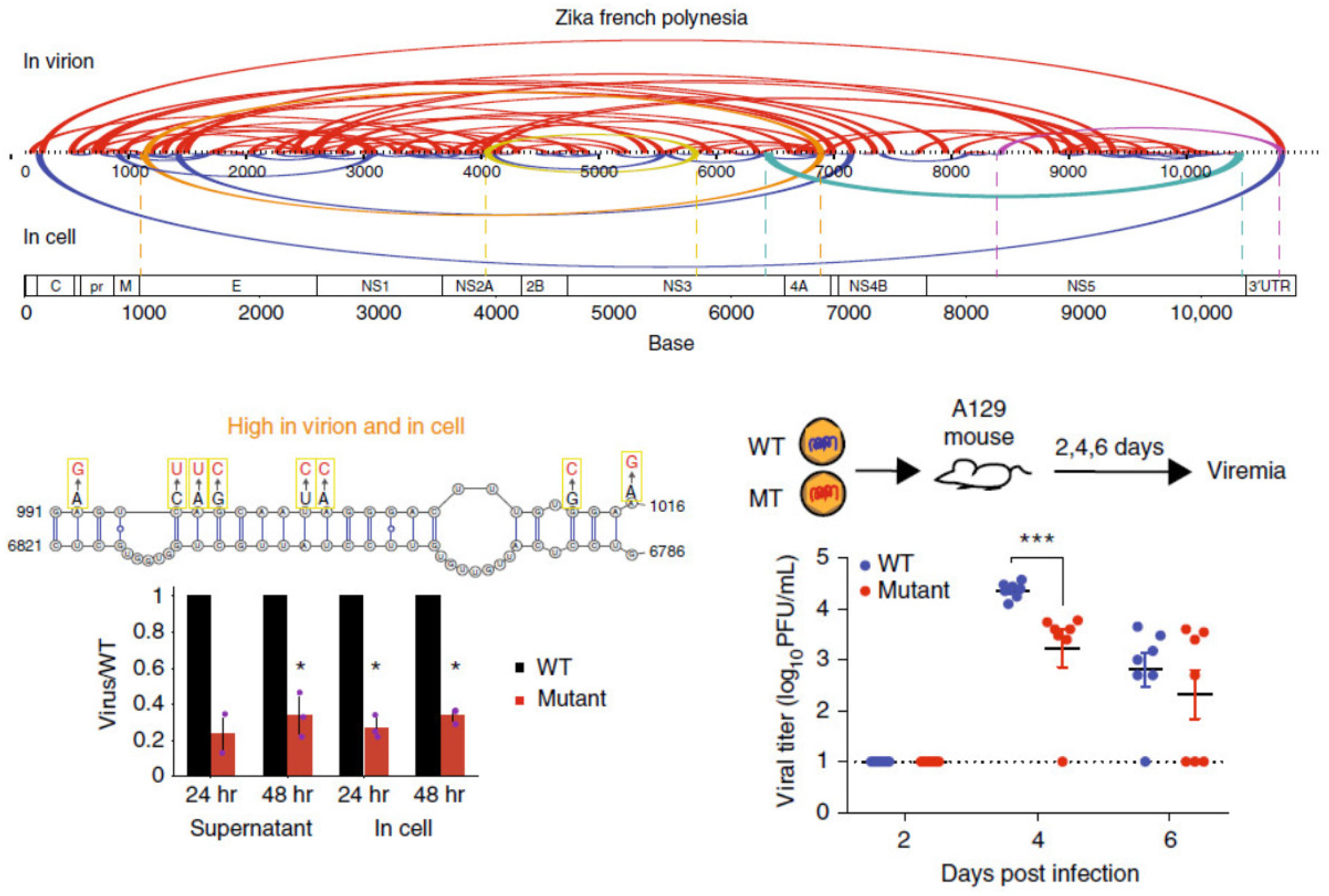{kind=link}
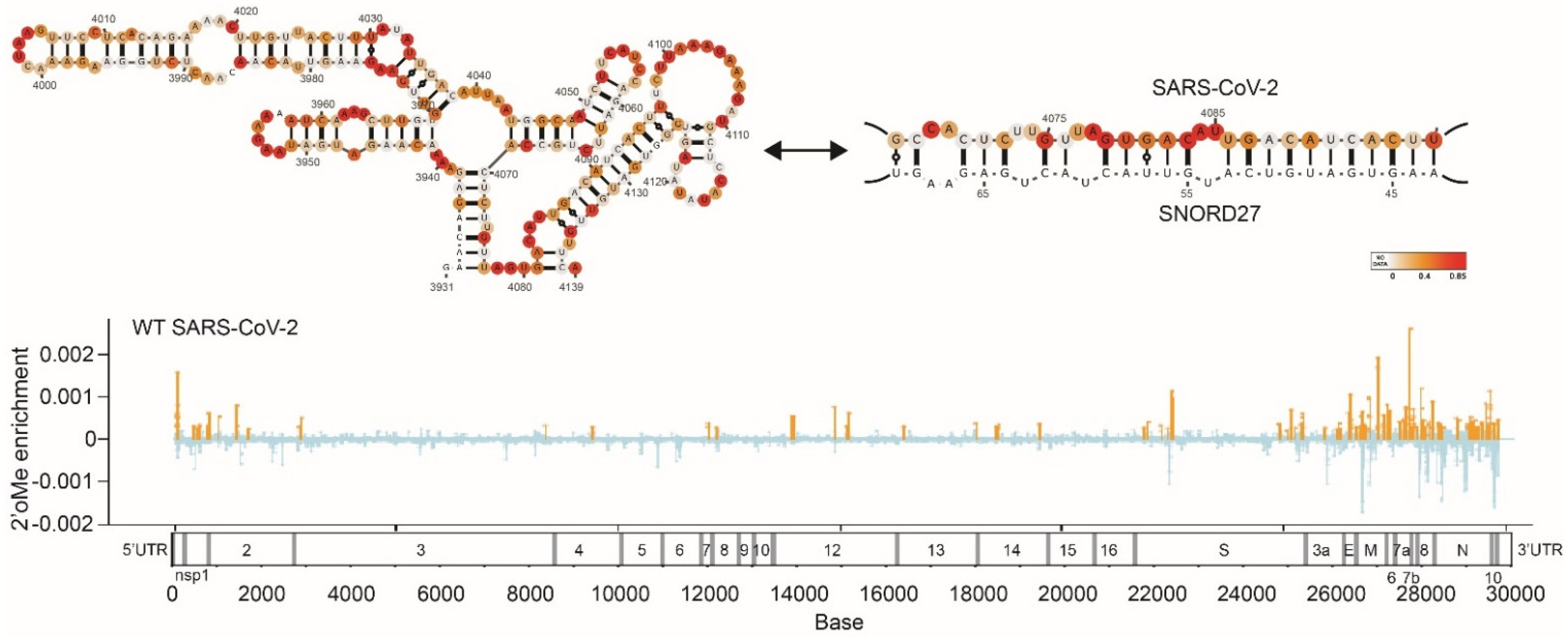
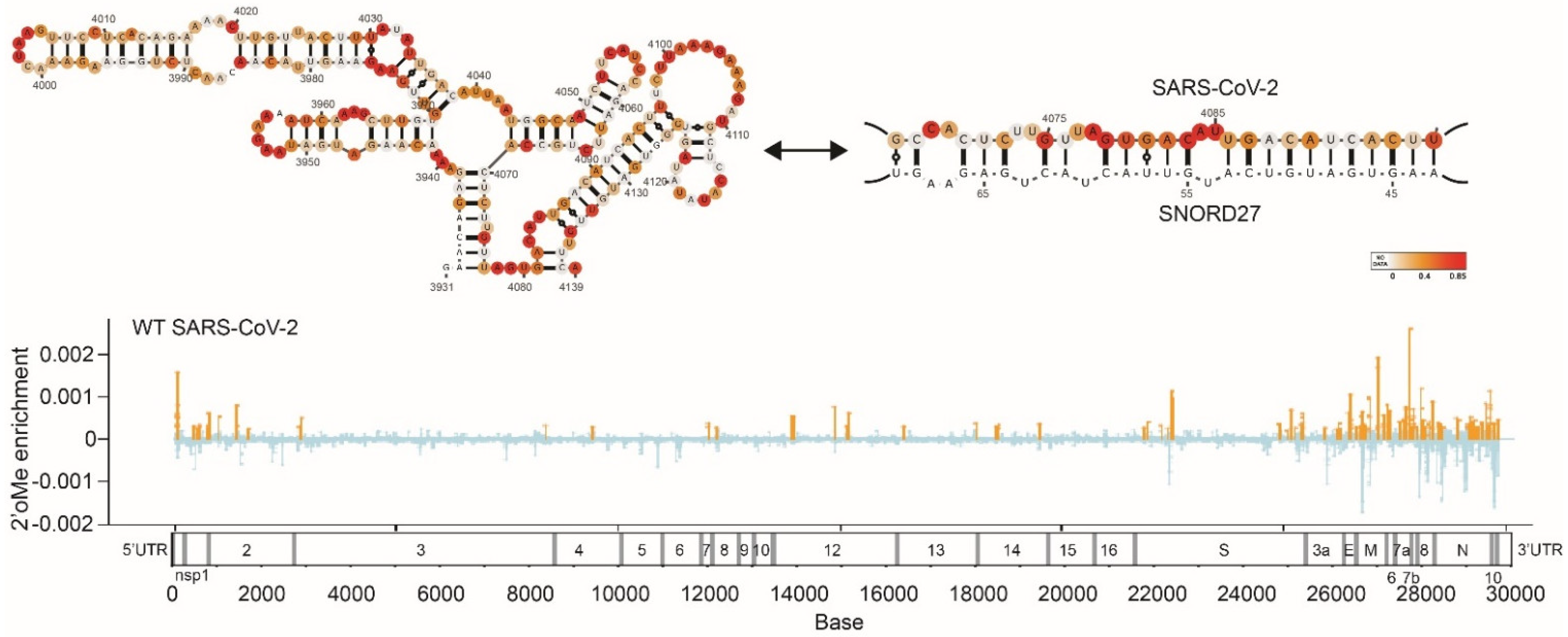{kind=link}
| Application | Methods | Algorithm Purpose | Input | References |
|---|---|---|---|---|
| Prediction of RNA secondary structure | ||||
| Thermodynamics-based | RNAfold, RNAstructure | Predicts the RNA secondary structure of a standalone sequence | RNA sequence | [20,21] |
| Comparative-based | RNAalifold, TurboFold, Dynalign, Multilign, FoldalignM | Predicts the RNA secondary structure using multiple sequences | Multiple-alignment/RNA sequence | [27,28,29,30,31] |
| AI-based | CROSS, ShaKer | Predicts the RNA secondary structure of a standalone sequence | RNA sequence | [58,59] |
| Combined approaches | RNAfold, RNAstructure, Superfold | Predicts the RNA secondary structure using experimental data as constraints | RNA sequence, SHAPE profile | [20,21,25] |
| Identification of functional RNA structures | ||||
| Secondary structure conservation | R-scape, RNA-Decoder | Identify covariation, base-pairing probability across many sequences | Multiple-alignment | [33,50] |
| Stability and potential functionality | ScanFold, RNAvigator | Identify RNA regions that are more experimentally stable than expected, identify regions of structural importance | RNA sequence, SHAPE profile | [47,51] |
| Method | Chemical Probe | Strategies | Advantages | Limitations | References |
|---|---|---|---|---|---|
| SHAPE-MaP | 1M7, NAI, 2A3 | Use SHAPE compounds to probe ssRNA regions. The mutations are detected through RT mutation read throughs | Probes all four nucleotides, analysis of low-abundance RNAs | Low jump through mutation rate, requires deep sequencing, no dsRNA information | [25,46,49,76,77] |
| icSHAPE | NAI-N3 | Probes ssRNA regions, biotin enrichment for modified fragments, RT-stop read out | Probes all four nucleotides, high signal-to-noise ratio | No dsRNA information | [36,64,78] |
| PARIS | AMT crosslinking | Psoralen-based crosslinking of dsRNAs, 2D gel extraction, proximity ligation and sequencing | Genome-wide in vivo RNA–RNA interactions, near base-pair resolution | Psoralen preferentially integrates into pyrimidine-rich sequences, proximity ligation in dilute solution | [39,64] |
| COMRADES | Psoralen-TEG-azide crosslinking | Psoralen-based crosslinking of dsRNAs, enrichment of RNA of interest using biotinylated probe, second biotin enrichment for crosslinked regions, proximity ligation and sequencing | Genome-wide in vivo RNA–RNA interactions of a specific RNA | Psoralen preferentially integrates into pyrimidine-rich sequence, proximity ligation in dilute solution | [40,79] |
| SPLASH | Biotinylated-psoralen | Psoralen-based crosslinking of dsRNAs, biotin enrichment for crosslinked regions, proximity ligation and sequencing | Genome-wide in vivo RNA–RNA interactions, high signal-to-noise ratio | Psoralen preferentially integrates into pyrimidine-rich sequence, proximity ligation in dilute solution | [34,38,49,80,81] |
| RING-MaP | DMS | DMS methylation on A and C, RT mutation read out | Structure probing of RNAs in 3D tertiary conformations | Only probes As and Cs, requires deep sequencing, and is mostly used for highly abundant RNAs | [65,82] |
| DMS-MaPseq | DMS | DMS methylation on A and C, RT mutation read out | Structure probing of RNAs in multiple conformations, analysis of low-abundance RNAs, high signal-to-noise ratio | Only probes As and Cs, no dsRNA information | [37,52,76] |
| PORE-cupine | NAI | Probes ssRNA regions, RT mutation read out using Nanopore full-length direct RNA sequencing | Probes all four nucleotides. Long-read sequencing enables capture of structural information of RNA isoforms and full-length transcripts | No dsRNA information, low sequencing depth. | [49,83] |
| vRIC-Seq | Formaldehyde crosslinking | In situ RNA digestion by nuclease, in situ proximity ligation, biotin enrichment for ligated fragments | Genome-wide in vivo RNA–RNA interactions, high signal-to-noise ratio, high percentage of chimeric reads | Formaldehyde crosslinking may introduce protein–protein, along with protein–RNA, interactions | [41,45] |
| Virus Family/Genus | Virus Species | Methods | Year | Reference |
|---|---|---|---|---|
| Retroviridae/Lentivirus | HIV-1 | SHAPE | 2009 | [35] |
| SIVmac239, HIV-1 | SHAPE | 2013 | [84] | |
| HIV-1 | SHAPE-MaP | 2014 | [25] | |
| SIVcpz, SIVmac, HIV-1 | SHAPE | 2015 | [85] | |
| Picornaviridae/Enterovirus | Poliovirus | SHAPE | 2013 | [86] |
| Flaviviridae/Hepacivirus | HCV | SHAPE-MaP | 2015 | [46] |
| HCV | SHAPE | 2016 | [87] | |
| Flaviviridae/Flavivirus | DENV2 | SHAPE-MaP, RING-MaP | 2018 | [65] |
| ZIKV | icSHAPE, PARIS | 2018 | [64] | |
| ZIKV | COMRADES | 2018 | [40] | |
| DENV1–4, ZIKV | NAI-MaP, SPLASH | 2019 | [34] | |
| Orthomyxoviridae/ Alphainfluenzavirus | IAV | SHAPE-MaP, SPLASH | 2019 | [80] |
| IAV | 2CIMPL | 2021 | [88] | |
| Coronaviridae/Betacoronavirus | SARS-CoV-2 | Nanopore DRS, DNBseq | 2020 | [89] |
| SARS-CoV-2 | SHAPE-MaP, DMS-MaPseq | 2020 | [76] | |
| SARS-CoV-2 | COMRADES | 2020 | [79] | |
| SARS-CoV-2 | icSHAPE | 2021 | [68] | |
| SARS-CoV-2 | SHAPE-MaP | 2021 | [48] | |
| SARS-CoV-2 | vRIC-seq | 2021 | [41] | |
| SARS-CoV-2 | SHAPE-MaP, PORE-cupine, SPLASH | 2021 | [49] | |
| SARS-CoV-2 | Simplified SPLASH | 2021 | [81] | |
| SARS-CoV-2 | DMS-MaPseq | 2022 | [52] | |
| SARS-CoV, MERS-CoV, SARS-CoV-2 | SHAPE-MaP | 2020 | [90] |
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Nachbagauer, R.; Palese, P. Is a universal influenza virus vaccine possible? Annu. Rev. Med. 2020, 71, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.C. Global burden of the HIV pandemic. Lancet 1996, 348, 99–106. [Google Scholar] [CrossRef]
- Garcia-Moreno, M.; Järvelin, A.I.; Castello, A. Unconventional RNA-binding proteins step into the virus-host battlefront. Wiley Interdiscip. Rev. RNA 2018, 9, e1498. [Google Scholar] [CrossRef] [PubMed]
- Embarc-Buh, A.; Francisco-Velilla, R.; Martinez-Salas, E. RNA-binding proteins at the host-pathogen interface targeting viral regulatory elements. Viruses 2021, 13, 952. [Google Scholar] [CrossRef]
- Boerneke, M.A.; Ehrhardt, J.E.; Weeks, K.M. Physical and functional analysis of viral RNA genomes by SHAPE. Annu. Rev. Virol. 2019, 6, 93–117. [Google Scholar] [CrossRef]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef]
- World Health Organization. Dengue Guidelines for Diagnosis, Treatment, Prevention and Control: New Edition; World Health Organization: Geneva, Switzerland, 2009. [Google Scholar]
- Junior, J.B.S.; Massad, E.; Lobao-Neto, A.; Kastner, R.; Oliver, L.; Gallagher, E. Epidemiology and costs of dengue in Brazil: A systematic literature review. Int. J. Infect. Dis. 2022, 122, 521–528. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Raveendran, A.V.; Jayadevan, R.; Sashidharan, S. Long COVID: An overview. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 869–875. [Google Scholar] [CrossRef]
- Steinman, M.; de Sousa, J.H.B.; Tustumi, F.; Wolosker, N. The burden of the pandemic on the non-SARS-CoV-2 emergencies: A multicenter study. Am. J. Emerg Med. 2021, 42, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Lukavsky, P.J. Structure and function of HCV IRES domains. Virus Res. 2009, 139, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Lukavsky, P.J.; Kim, I.; Otto, G.A.; Puglisi, J.D. Structure of HCV IRES domain II determined by NMR. Nat. Struct. Biol. 2003, 10, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Beckham, S.A.; Matak, M.Y.; Belousoff, M.J.; Venugopal, H.; Shah, N.; Vankadari, N.; Elmlund, H.; Nguyen, J.H.C.; Semler, B.L.; Wilce, M.C.J.; et al. Structure of the PCBP2/stem-loop IV complex underlying translation initiation mediated by the poliovirus type I IRES. Nucleic Acids Res. 2020, 48, 8006–8021. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.R.; Sarnow, P. Enterovirus 71 contains a type I IRES element that functions when eukaryotic initiation factor eIF4G is cleaved. Virology 2003, 315, 259–266. [Google Scholar] [CrossRef]
- Brierley, I.; Digard, P.; Inglis, S.C. Characterization of an efficient coronavirus ribosomal frameshifting signal: Requirement for an RNA pseudoknot. Cell 1989, 57, 537–547. [Google Scholar] [CrossRef]
- Paillart, J.C.; Skripkin, E.; Ehresmann, B.; Ehresmann, C.; Marquet, R. In vitro evidence for a long range pseudoknot in the 5′-untranslated and matrix coding regions of HIV-1 genomic RNA. J. Biol. Chem. 2002, 277, 5995–6004. [Google Scholar] [CrossRef]
- Gebhard, L.G.; Filomatori, C.V.; Gamarnik, A.V. Functional RNA elements in the dengue virus genome. Viruses 2011, 3, 1739–1756. [Google Scholar] [CrossRef]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef]
- Martin, F.H.; Uhlenbeck, O.C.; Doty, P. Self-complementary oligoribonucleotides: Adenylic acid-uridylic acid block copolymers. J. Mol. Biol. 1971, 57, 201–215. [Google Scholar] [CrossRef]
- Hajdin, C.E.; Bellaousov, S.; Huggins, W.; Leonard, C.W.; Mathews, D.H.; Weeks, K.M. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc. Natl Acad. Sci. USA 2013, 110, 5498–5503. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.J.; Gloor, J.W.; Mathews, D.H. Improved RNA secondary structure prediction by maximizing expected pair accuracy. RNA 2009, 15, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, N.A.; Busan, S.; Rice, G.M.; Nelson, J.A.E.; Weeks, K.M. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 2014, 11, 959–965. [Google Scholar] [CrossRef]
- Bokov, K.; Steinberg, S.V. A hierarchical model for evolution of 23S ribosomal RNA. Nature 2009, 457, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, S.H.; Hofacker, I.L.; Will, S.; Gruber, A.R.; Stadler, P.F. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinform. 2008, 9, 474. [Google Scholar] [CrossRef] [PubMed]
- Harmanci, A.O.; Sharma, G.; Mathews, D.H. TurboFold: Iterative probabilistic estimation of secondary structures for multiple RNA sequences. BMC Bioinform. 2011, 12, 108. [Google Scholar] [CrossRef]
- Fu, Y.; Sharma, G.; Mathews, D.H. Dynalign II: Common secondary structure prediction for RNA homologs with domain insertions. Nucleic Acids Res. 2014, 42, 13939–13948. [Google Scholar] [CrossRef]
- Xu, Z.; Mathews, D.H. Multilign: An algorithm to predict secondary structures conserved in multiple RNA sequences. Bioinformatics 2011, 27, 626–632. [Google Scholar] [CrossRef]
- Torarinsson, E.; Havgaard, J.H.; Gorodkin, J. Multiple structural alignment and clustering of RNA sequences. Bioinformatics 2007, 23, 926–932. [Google Scholar] [CrossRef]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- Rivas, E.; Clements, J.; Eddy, E.R.S.R. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat. Methods 2017, 14, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.G.; Lim, X.N.; Ng, W.C.; Sim, A.Y.L.; Poh, H.X.; Shen, Y.; Lim, S.Y.; Sundstrom, K.B.; Sun, X.; Aw, J.G.; et al. Structure mapping of dengue and Zika viruses reveals functional long-range interactions. Nat. Commun. 2019, 10, 1408. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.M.; Dang, K.K.; Gorelick, R.J.; Leonard, C.W.; Bess Jr, J.W.; Swanstrom, R.; Burch, C.L.; Weeks, K.M. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009, 460, 711–716. [Google Scholar] [CrossRef]
- Spitale, R.C.; Flynn, R.; Zhang, Q.C.; Crisalli, P.; Lee, B.; Jung, J.W.; Kuchelmeister, H.Y.; Batista, P.J.; Torre, E.A.; Kool, E.T.; et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 2015, 519, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Zubradt, M.; Gupta, P.; Persad, S.; Lambowitz, A.M.; Weissman, J.S.; Rouskin, S. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 2017, 14, 75–82. [Google Scholar] [CrossRef]
- Aw, J.G.A.; Shen, Y.; Wilm, A.; Sun, M.; Lim, X.N.; Boon, K.L.; Tapsin, S.; Chan, Y.S.; Tan, C.P.; Sim, A.Y.; et al. In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol. Cell 2016, 62, 603–617. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, Q.C.; Lee, B.; Flynn, R.A.; Smith, M.A.; Robinson, J.T.; Davidovich, C.; Gooding, A.R.; Goodrich, K.J.; Mattick, J.S.; et al. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell 2016, 165, 1267–1279. [Google Scholar] [CrossRef]
- Ziv, O.; Gabryelska, M.M.; Lun, A.T.L.; Gebert, L.F.R.; Sheu-Gruttadauria, J.; Meredith, L.W.; Liu, Z.Y.; Kwok, C.K.; Qin, C.F.; MacRae, I.J.; et al. COMRADES determines in vivo RNA structures and interactions. Nat. Methods 2018, 15, 785–788. [Google Scholar] [CrossRef]
- Cao, C.; Cai, Z.; Xiao, X.; Rao, J.; Chen, J.; Hu, N.; Yang, M.; Xing, X.; Wang, Y.; Li, M.; et al. The architecture of the SARS-CoV-2 RNA genome inside virion. Nat. Commun. 2021, 12, 3917. [Google Scholar] [CrossRef]
- Strobel, E.J.; Yu, A.M.; Lucks, J.B. High-throughput determination of RNA structures. Nat. Rev. Genet. 2018, 19, 615–634. [Google Scholar] [CrossRef] [PubMed]
- Tomezsko, P.J.; Corbin, V.D.A.; Gupta, P.; Swaminathan, H.; Glasgow, M.; Persad, S.; Edwards, M.D.; McIntosh, L.; Papenfuss, A.T.; Emery, A.; et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 2020, 582, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Sharma, E.; Sterne-Weiler, T.; O’Hanlon, D.; Blencowe, B.J. Global mapping of human RNA-RNA interactions. Mol. cell 2016, 62, 618–626. [Google Scholar] [CrossRef]
- Cai, Z.; Cao, C.; Ji, L.; Ye, R.; Wang, D.; Xia, C.; Wang, S.; Du, Z.; Hu, N.; Yu, X.; et al. RIC-seq for global in situ profiling of RNA-RNA spatial interactions. Nature 2020, 582, 432–437. [Google Scholar] [CrossRef]
- Mauger, D.M.; Golden, M.; Yamane, D.; Williford, S.; Lemon Stanley, M.; Martin, D.P.; Weeks, K.M. Functionally conserved architecture of hepatitis C virus RNA genomes. Proc. Natl. Acad. Sci. USA 2015, 112, 3692–3697. [Google Scholar] [CrossRef]
- Andrews, R.J.; Roche, J.; Moss, W.N. ScanFold: An approach for genome-wide discovery of local RNA structural elements-applications to Zika virus and HIV. PeerJ 2018, 6, e6136. [Google Scholar] [CrossRef]
- Huston, N.C.; Wan, H.; Strine, M.S.; Tavares, R.D.C.A.; Wilen, C.B.; Pyle, A.M. Comprehensive in vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms. Mol. Cell 2021, 81, 584–598.e5. [Google Scholar] [CrossRef]
- Yang, S.L.; DeFalco, L.; Anderson, D.E.; Zhang, Y.; Aw, J.G.A.; Lim, S.Y.; Lim, X.N.; Tan, K.Y.; Zhang, T.; Chawla, T.; et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nat. Commun. 2021, 12, 5113. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Meyer, I.M.; Forsberg, R.; Simmonds, P.; Hein, J. A comparative method for finding and folding RNA secondary structures within protein-coding regions. Nucleic Acids Res. 2004, 32, 4925–4936. [Google Scholar] [CrossRef]
- Delli Ponti, R.; Wang, J.; Wan, Y.; Huber, R.G. RNAvigator: A Pipeline to Identify Candidates for Functional RNA Structure Elements. Front. Virol. 2022, 2, 878679. [Google Scholar] [CrossRef]
- Lan, T.C.; Allan, M.F.; Malsick, L.E.; Woo, J.Z.; Zhu, C.; Zhang, F.; Khandwala, S.; Nyeo, S.S.Y.; Sun, Y.; Guo, J.U.; et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nat. Commun. 2022, 13, 1128. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Luntzer, D.; Hofacker, I.L.; Stadler, P.F.; Wolfinger, M.T. SHAPE directed RNA folding. Bioinformatics 2016, 32, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Low, J.T.; Weeks, K.M. SHAPE-directed RNA secondary structure prediction. Methods 2010, 52, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Deigan, K.E.; Li, T.W.; Mathews, D.H.; Weeks, K.M. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. USA 2009, 106, 97–102. [Google Scholar] [CrossRef]
- Zarringhalam, K.; Meyer, M.M.; Dotu, I.; Chuang, J.H.; Clote, P. Integrating chemical footprinting data into RNA secondary structure prediction. PloS ONE 2012, 7, e45160. [Google Scholar] [CrossRef]
- Washietl, S.; Hofacker, I.L.; Stadler, P.F.; Kellis, M. RNA folding with soft constraints: Reconciliation of probing data and thermodynamic secondary structure prediction. Nucleic Acids Res. 2012, 40, 4261–4272. [Google Scholar] [CrossRef]
- Delli Ponti, R.; Marti, S.; Armaos, A.; Tartaglia, G.G. A high-throughput approach to profile RNA structure. Nucleic Acids Res. 2017, 45, e35. [Google Scholar] [CrossRef]
- Mautner, S.; Montaseri, S.; Miladi, M.; Raden, M.; Costa, F.; Backofen, R. ShaKer: RNA SHAPE prediction using graph kernel. Bioinformatics 2019, 35, i354–i359. [Google Scholar] [CrossRef]
- Vandelli, A.; Monti, M.; Milanetti, E.; Armaos, A.; Rupert, J.; Zacco, E.; Bechara, E.; Delli Ponti, R.; Tartaglia, G.G. Structural analysis of SARS-CoV-2 genome and predictions of the human interactome. Nucleic Acids Res. 2020, 48, 11270–11283. [Google Scholar] [CrossRef]
- Delli Ponti, R.; Mutwil, M. Structural landscape of the complete genomes of dengue virus serotypes and other viral hemorrhagic fevers. BMC Genom. 2021, 22, 352. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed]
- De Falco, L.; Silva, N.M.; Santos, N.C.; Huber, R.G.; Martins, I.C. The pseudo-circular genomes of flaviviruses: Structures, mechanisms, and functions of circularization. Cells 2021, 10, 642. [Google Scholar] [CrossRef]
- Li, P.; Wei, Y.; Mei, M.; Tang, L.; Sun, L.; Huang, W.; Zhou, J.; Zou, C.; Zhang, S.; Qin, C.F.; et al. Integrative analysis of Zika virus genome RNA structure reveals critical determinants of viral infectivity. Cell Host Microbe. 2018, 24, 875–886.e5. [Google Scholar] [CrossRef] [PubMed]
- Dethoff, E.A.; Boerneke, M.A.; Gokhale, N.S.; Muhire, B.M.; Martin, D.P.; Sacco, M.T.; McFadden, M.J.; Weinstein, J.B.; Messer, W.B.; Horner, S.M.; et al. Pervasive tertiary structure in the dengue virus RNA genome. Proc. Natl. Acad. Sci. USA 2018, 115, 11513–11518. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Flynn, R.A.; Belk, J.A.; Qi, Y.; Yasumoto, Y.; Wei, J.; Alfajaro, M.M.; Shi, Q.; Mumbach, M.R.; Limaye, A.; DeWeirdt, P.C.; et al. Discovery and functional interrogation of SARS-CoV-2 RNA-host protein interactions. Cell 2021, 184, 2394–2411.e16. [Google Scholar] [CrossRef]
- Sun, L.; Li, P.; Ju, X.; Rao, J.; Huang, W.; Ren, L.; Zhang, S.; Xiong, T.; Xu, K.; Zhou, X.; et al. In vivo structural characterization of the SARS-CoV-2 RNA genome identifies host proteins vulnerable to repurposed drugs. Cell 2021, 184, 1865–1883.e20. [Google Scholar] [CrossRef]
- Haniff, H.S.; Tong, Y.; Liu, X.; Chen, J.L.; Suresh, B.M.; Andrews, R.J.; Peterson, J.M.; O’Leary, C.A.; Benhamou, R.I.; Moss, W.N.; et al. Targeting the SARS-CoV-2 RNA genome with small molecule binders and ribonuclease targeting chimera (RIBOTAC) degraders. ACS Cent. Sci. 2020, 6, 1713–1721. [Google Scholar] [CrossRef]
- Sreeramulu, S.; Richter, C.; Berg, H.; Wirtz Martin, M.A.; Ceylan, B.; Matzel, T.; Adam, J.; Altincekic, N.; Azzaoui, K.; Bains, J.K.; et al. Exploring the druggability of conserved RNA regulatory elements in the SARS-CoV-2 genome. Angew. Chem. Int. Ed. 2021, 60, 19191–19200. [Google Scholar] [CrossRef]
- Morandi, E.; Manfredonia, I.; Simon, L.M.; Anselmi, F.; van Hemert, M.J.; Oliviero, S.; Incarnato, D. Genome-scale deconvolution of RNA structure ensembles. Nat. Methods 2021, 18, 249–252. [Google Scholar] [CrossRef]
- Stelzer, A.C.; Frank, A.T.; Kratz, J.D.; Swanson, M.D.; Gonzalez-Hernandez, M.J.; Lee, J.; Andricioaei, I.; Markovitz, D.M.; Al-Hashimi, H.M. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 2011, 7, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Jia, X.; Zhang, K.; Su, Z. Cryo-EM advances in RNA structure determination. Signal Transduct. Target. 2022, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zheludev, I.N.; Hagey, R.J.; Haslecker, R.; Hou, Y.J.; Kretsch, R.; Pintilie, G.D.; Rangan, R.; Kladwang, W.; Li, S.; et al. Cryo-EM and antisense targeting of the 28-kDa frameshift stimulation element from the SARS-CoV-2 RNA genome. Nat. Struct. Mol. Biol. 2021, 28, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-T.; Varani, G. Structure of the Dengue Virus RNA Promoter. bioRxiv 2022, 2022.04.15.488410. [Google Scholar] [CrossRef]
- Manfredonia, I.; Nithin, C.; Ponce-Salvatierra, A.; Ghosh, P.; Wirecki, T.K.; Marinus, T.; Ogando, N.S.; Snijder, E.J.; van Hemert, M.J.; Bujnicki, J.M.; et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Res. 2020, 48, 12436–12452. [Google Scholar] [CrossRef]
- Marinus, T.; Fessler, A.B.; Ogle, C.A.; Incarnato, D. A novel SHAPE reagent enables the analysis of RNA structure in living cells with unprecedented accuracy. Nucleic Acids Res. 2021, 49, e34. [Google Scholar] [CrossRef]
- Flynn, R.A.; Zhang, Q.C.; Spitale, R.C.; Lee, B.; Mumbach, M.R.; Chang, H.Y. Transcriptome-wide interrogation of RNA secondary structure in living cells with icSHAPE. Nat. Protoc. 2016, 11, 273–290. [Google Scholar] [CrossRef]
- Ziv, O.; Price, J.; Shalamova, L.; Kamenova, T.; Goodfellow, I.; Weber, F.; Miska, E.A. The short- and long-range RNA-RNA interactome of SARS-CoV-2. Mol. Cell 2020, 80, 1067–1077.e5. [Google Scholar] [CrossRef]
- Dadonaite, B.; Gilbertson, B.; Knight, M.L.; Trifkovic, S.; Rockman, S.; Laederach, A.; Brown, L.E.; Fodor, E.; Bauer, D.L.V. The structure of the influenza A virus genome. Nat. Microb. 2019, 4, 1781–1789. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, K.; Xie, D.; Lau, J.Y.; Shen, W.; Li, P.; Wang, D.; Zou, Z.; Shi, S.; Ren, H.; et al. In vivo structure and dynamics of the SARS-CoV-2 RNA genome. Nat. Commun. 2021, 12, 5695. [Google Scholar] [CrossRef]
- Homan, P.J.; Favorov, O.V.; Lavender, C.A.; Kursun, O.; Ge, X.; Busan, S.; Dokholyan, N.V.; Weeks, K.M. Single-molecule correlated chemical probing of RNA. Proc. Natl. Acad. Sci. USA 2014, 111, 13858–13863. [Google Scholar] [CrossRef] [PubMed]
- Aw, J.G.A.; Lim, S.W.; Wang, J.X.; Lambert, F.R.P.; Tan, W.T.; Shen, Y.; Zhang, Y.; Kaewsapsak, P.; Li, C.; Ng, S.B.; et al. Determination of isoform-specific RNA structure with nanopore long reads. Nat. Biotech. 2021, 39, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Pollom, E.; Dang, K.K.; Potter, E.L.; Gorelick, R.J.; Burch, C.L.; Weeks, K.M.; Swanstrom, R. Comparison of SIV and HIV-1 genomic RNA structures reveals impact of sequence evolution on conserved and non-conserved structural motifs. PLoS Pathog. 2013, 9, e1003294. [Google Scholar] [CrossRef] [PubMed]
- Lavender, C.A.; Gorelick, R.J.; Weeks, K.M. Structure-Based Alignment and Consensus Secondary Structures for Three HIV-Related RNA Genomes. PLOS Comput. Biol. 2015, 11, e1004230. [Google Scholar] [CrossRef]
- Burrill, C.P.; Westesson, O.; Schulte, M.B.; Strings, V.R.; Segal, M.; Andino, R. Global RNA structure analysis of poliovirus identifies a conserved RNA structure involved in viral replication and infectivity. J. Virol. 2013, 87, 11670–11683. [Google Scholar] [CrossRef]
- Pirakitikulr, N.; Kohlway, A.; Lindenbach, B.D.; Pyle, A.M. The coding region of the HCV genome contains a network of regulatory RNA structures. Mol. Cell 2016, 62, 111–120. [Google Scholar] [CrossRef]
- Le Sage, V.; Kanarek, J.P.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S.; Lee, N. Mapping of influenza virus RNA-RNA interactions reveals a flexible network. Cell Rep. 2020, 31, 107823. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Sanders, W.; Fritch, E.J.; Madden, E.A.; Graham, R.L.; Vincent, H.A.; Heise, M.T.; Baric, R.S.; Moorman, N.J. Comparative analysis of coronavirus genomic RNA structure reveals conservation in SARS-like coronaviruses. Biorxiv Prepr. Serv. Biol. 2020, 2020.06.15.153197. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.L.; Ponti, R.D.; Wan, Y.; Huber, R.G. Computational and Experimental Approaches to Study the RNA Secondary Structures of RNA Viruses. Viruses 2022, 14, 1795. https://doi.org/10.3390/v14081795
Yang SL, Ponti RD, Wan Y, Huber RG. Computational and Experimental Approaches to Study the RNA Secondary Structures of RNA Viruses. Viruses. 2022; 14(8):1795. https://doi.org/10.3390/v14081795
Chicago/Turabian StyleYang, Siwy Ling, Riccardo Delli Ponti, Yue Wan, and Roland G. Huber. 2022. "Computational and Experimental Approaches to Study the RNA Secondary Structures of RNA Viruses" Viruses 14, no. 8: 1795. https://doi.org/10.3390/v14081795
APA StyleYang, S. L., Ponti, R. D., Wan, Y., & Huber, R. G. (2022). Computational and Experimental Approaches to Study the RNA Secondary Structures of RNA Viruses. Viruses, 14(8), 1795. https://doi.org/10.3390/v14081795