
Viral Nucleases from Herpesviruses and Coronavirus in Recombination and Proofreading: Potential Targets for Antiviral Drug Discovery

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. General Mechanism of Recombination
3. Human Herpesviruses (HHVs)
3.1. Primary Infection
3.2. Latent Infection
3.3. Possible Association of HHVs with Neurodegenerative Diseases
3.4. Replication Strategies of the HHVs
3.5. Current Standard of Care
4. Introduction to Coronaviruses
4.1. Role of Recombination in CoV Infection
4.2. General Aspects of CoV Replication
4.3. CoV Replicase Proteins
4.4. Identification of ExoN as a Driver for Recombination
4.5. Current Standard of Care
4.6. Similarities between HHVs and CoVs
5. Introduction to TMID Nucleases
6. Exonuclease Enzymes of HHV
6.1. Alkaline Nuclease
6.2. PolExo
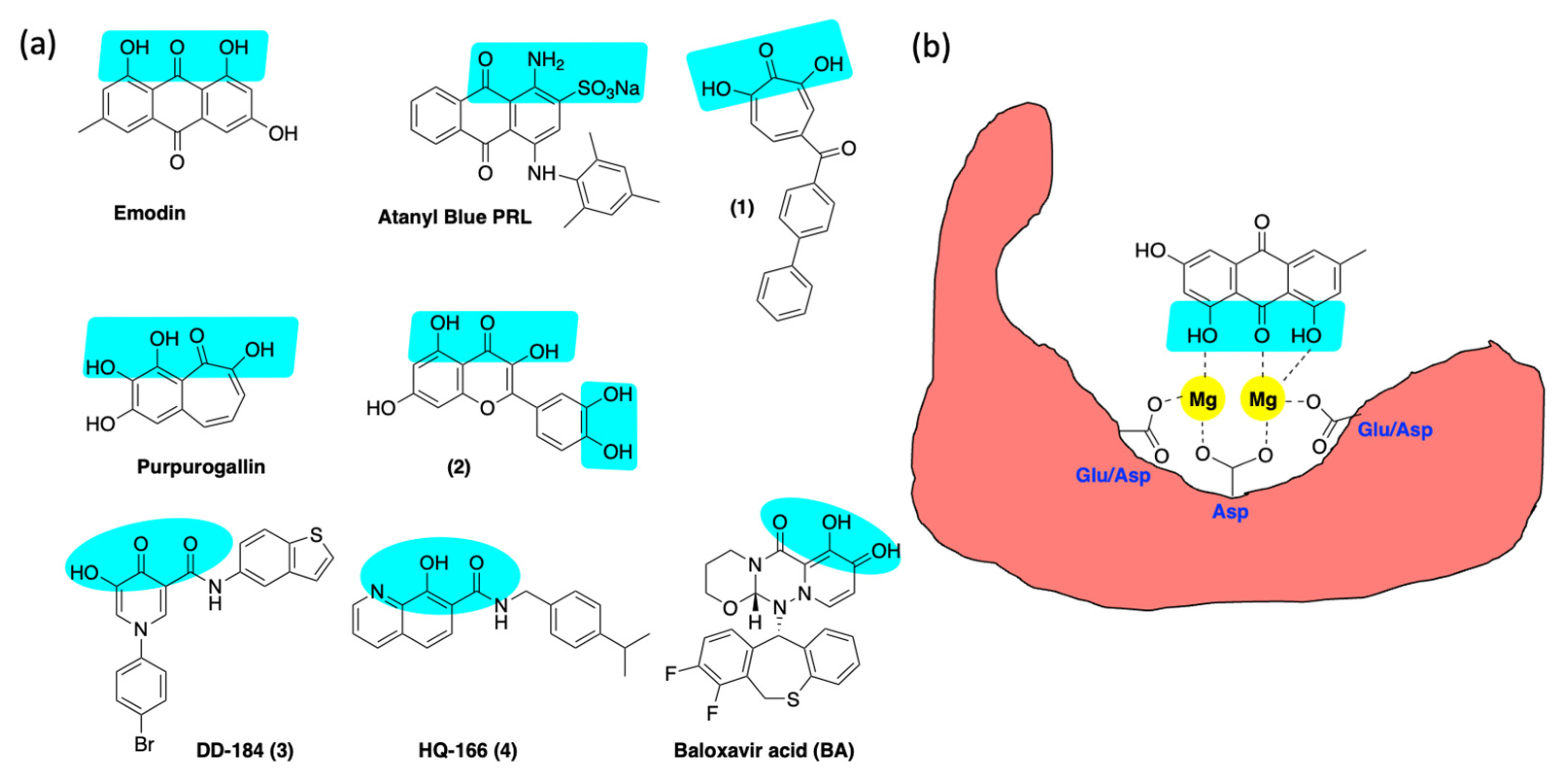
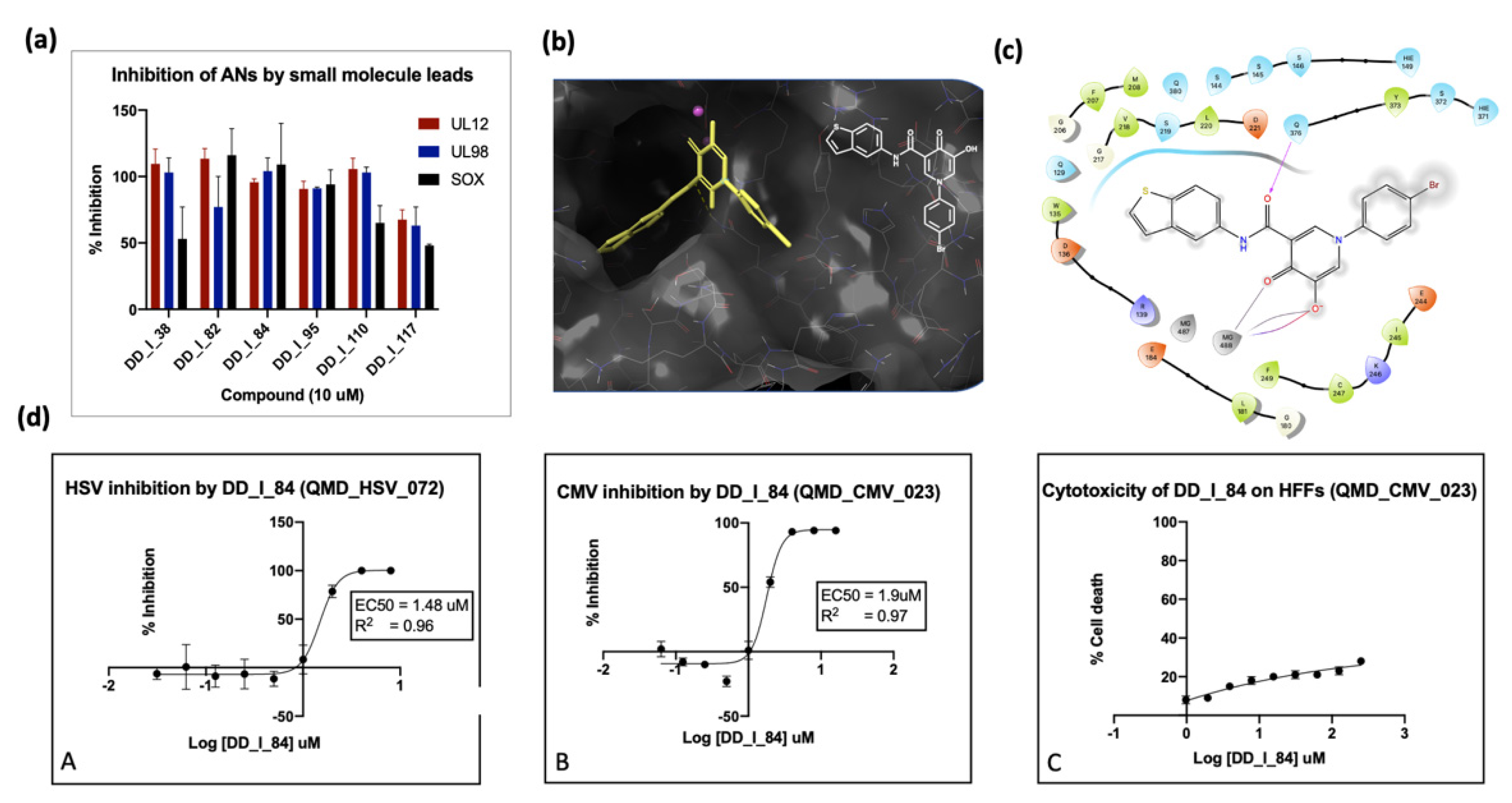
6.3. Inhibition of Alkaline Nuclease and Pol/Exo Function
7. nsp14/nsp10 Proofreading Exonuclease of Coronaviruses
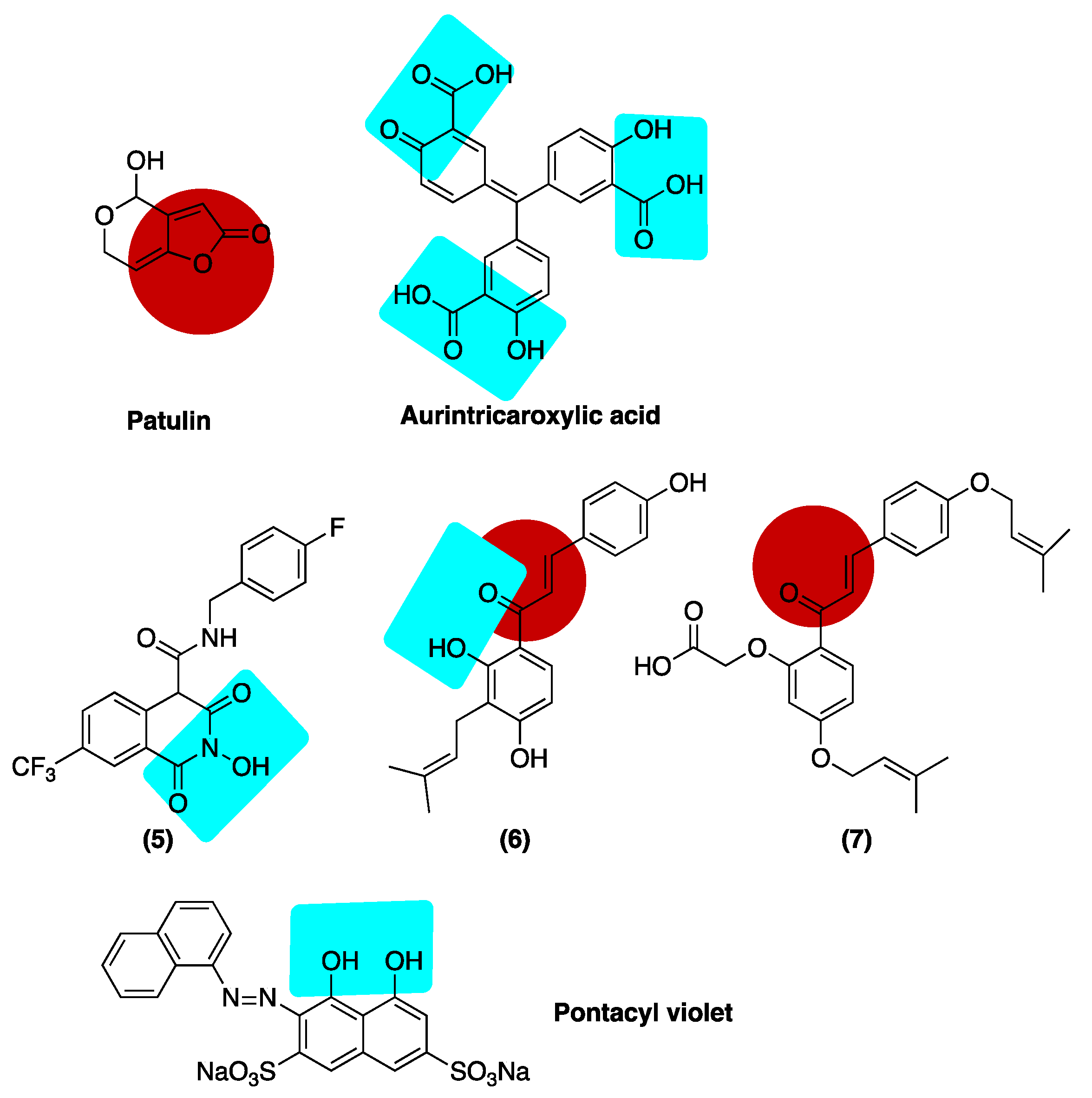
Inhibition of ExoN
8. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mosig, G.; Gewin, J.; Luder, A.; Colowick, N.; Vo, D. Two Recombination-Dependent DNA Replication Pathways of Bacteriophage T4, and Their Roles in Mutagenesis and Horizontal Gene Transfer. Proc. Natl. Acad. Sci. USA 2001, 98, 8306–8311. [Google Scholar] [CrossRef] [PubMed]
- Brewster, J.L.; Tolun, G. Half a Century of Bacteriophage Lambda Recombinase: In Vitro Studies of Lambda Exonuclease and Red-Beta Annealase. IUBMB Life 2020, 72, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.K.; Coen, D.M. Herpes Simplex Viruses: Mechanisms of DNA Replication. Cold Spring Harb. Perspect. Biol. 2012, 4, 4:a013011. [Google Scholar] [CrossRef]
- Schumacher, A.; Mohni, K.; Kan, Y.; Hendrickson, E.; Stark, J.; Weller, S.K. The HSV-1 Exonuclease, UL12, Stimulates Recombination by a Single Strand Annealing Mechanism. PLoS Pathog. 2012, 8, e1002862. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.K.; Sawitzke, J.A. Recombination Promoted by DNA Viruses: Phage λ to Herpes Simplex Virus. Annu. Rev. Microbiol 2014, 68, 237–258. [Google Scholar] [CrossRef]
- Weerasooriya, S.; DiScipio, K.A.; Darwish, A.S.; Bai, P.; Weller, S.K. Herpes Simplex Virus 1 ICP8 Mutant Lacking Annealing Activity Is Deficient for Viral DNA Replication. Proc. Natl. Acad. Sci. USA 2019, 116, 1033–1042. [Google Scholar] [CrossRef]
- Lowen, A.C. Constraints, Drivers, and Implications of Influenza A Virus Reassortment. Annu. Rev. Virol. 2017, 4, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D.D.; Douglass Wright, W.; Shital Shah, S.; Heyer, W.-D.D. Homologous Recombination and the Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Symington, L.S. Mechanism and Regulation of DNA End Resection in Eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef]
- Reddy, G.; Golub, E.I.; Radding, C.M. Human Rad52 Protein Promotes Single-Strand DNA Annealing Followed by Branch Migration. Mutat. Res. - Fundam. Mol. Mech. Mutagen. 1997, 377, 53–59. [Google Scholar] [CrossRef]
- Brandsma, I.; Gent, D.C. Pathway Choice in DNA Double Strand Break Repair: Observations of a Balancing Act. Genome Integr. 2012, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.; Rudd, K.E. Mining DNA Sequences for Molecular Enzymology: The Red Alpha Superfamily Defines a Set of Recombination Nucleases. Proc. 1998 Miami Nat. Biotchnology Winter Symp. 1998, 38, 49–50. [Google Scholar]
- Aravind, L.; Makarova, K.S.; Koonin, E.V. Holliday Junction Resolvases and Related Nucleases: Identification of New Families, Phyletic Distribution and Evolutionary Trajectories. Nucleic Acids Res. 2000, 28, 3417–3432. [Google Scholar] [CrossRef]
- Reuven, N.; Staire, A.E.; Myers, R.; Weller, S.K. The Herpes Simplex Virus Type 1 Alkaline Nuclease and Single-Stranded DNA Binding Protein Mediate Strand Exchange In Vitro. J. Virol. 2003, 77, 7425–7433. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.; Willcox, S.; Griffith, J.D.; Weller, S.K. Catalysis of Strand Exchange by the HSV-1 UL12 and ICP8 Proteins: Potent ICP8 Recombinase Activity Is Revealed upon Resection of DsDNA Substrate by Nuclease. J. Mol. Biol. 2004, 342, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, V.S.; Okano, K.; Rohrmann, G.F. Baculovirus Alkaline Nuclease Possesses a 5′→3′ Exonuclease Activity and Associates with the DNA-Binding Protein LEF-3. J. Virol. 2003, 77, 2436–2444. [Google Scholar] [CrossRef] [PubMed]
- Fanning, E.; Zhao, K. SV40 DNA Replication: From the A Gene to a Nanomachine. Virology 2009, 384, 352–359. [Google Scholar] [CrossRef]
- Court, D.L.; Sawitzke, J.A.; Thomason, L.C. Genetic Engineering Using Homologous Recombination. Annu. Rev. Genet. 2002, 36, 361–388. [Google Scholar] [CrossRef]
- Sawitzke, J.A.; Thomason, L.C.; Costantino, N.; Bubunenko, M.; Datta, S.; Court, D.L. Recombineering: In Vivo Genetic Engineering in E. Coli, S. Enterica, and Beyond. Methods Enzymol. 2007, 421, 171–199. [Google Scholar] [CrossRef]
- Grady, L.; Szczepaniak, R.; Murelli, R.P.; Masaoka, T.; Le Grice, S.F.J.; Wright, D.; Weller, S.K. The Exonuclease Activity of Herpes Simplex Virus 1 UL12 Is Required for Production of Viral DNA That Can Be Packaged To Produce Infectious Virus. J. Virol. 2017, 91, e01380-17. [Google Scholar] [CrossRef]
- Darwish, A.S.; Grady, L.; Bai, P.; Weller, S.K. ICP8 Filament Formation Is Essential for Replication Compartment Formation during Herpes Simplex Virus Infection. J. Virol. 2016, 90, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Chayavichitsilp, P.; Buckwalter, J.V.; Krakowski, A.C.; Friedlander, S.F. Herpes Simplex. Pediatr. Rev. 2009, 30, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Dollard, S.C.; Grosse, S.D.; Ross, D.S. New Estimates of the Prevalence of Neurological and Sensory Sequelae and Mortality Associated with Congenital Cytomegalovirus Infection. Rev. Med. Virol. 2007, 17, 355–363. [Google Scholar] [CrossRef]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “Silent” Global Burden of Congenital Cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Heiden, D.; Ford, N.; Wilson, D.; Rodriguez, W.R.; Morgolis, T.; Janssens, B.; Bedelu, M.; Tun, N.; Geomaere, E.; Sranchuk, P.; et al. Cyomegalovirus Retinitis: The Neglected Disease of the AIDS Pandemic. PLoS Med. 2007, 4, 1845–1851. [Google Scholar] [CrossRef]
- Sinclair, J.; Sissons, P. Latency and Reactivation of Human Cytomegalovirus. J. Gen. Virol. 2006, 87, 1763–1779. [Google Scholar] [CrossRef] [PubMed]
- Harberts, E.; Yao, K.; Wohler, J.E.; Maric, D.; Ohayon, J.; Henkin, R.; Jacobson, S. Human Herpesvirus-6 Entry into the Central Nervous System through the Olfactory Pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 13734–13739. [Google Scholar] [CrossRef] [PubMed]
- Komaroff, A.L.; Pellett, P.E.; Jacobson, S. Human Herpesviruses 6a and 6b in Brain Diseases: Association versus Causation. Clin. Microbiol. Rev. 2021, 34, 1–36. [Google Scholar] [CrossRef]
- Bharucha, T.; Houlihan, C.F.; Breuer, J. Herpesvirus Infections of the Central Nervous System. Semin. Neurol. 2019, 39, 369–382. [Google Scholar] [CrossRef]
- Cohen, J.I. Herpesvirus Latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef]
- Abad, C.L.; Razonable, R.R. Treatment of Alpha and Beta Herpesvirus Infections in Solid Organ Transplant Recipients. Expert Rev. Anti. Infect. Ther. 2017, 15, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Meesing, A.; Razonable, R.R. Pharmacologic and Immunologic Management of Cytomegalovirus Infection after Solid Organ and Hematopoietic Stem Cell Transplantation. Expert Rev. Clin. Pharmacol. 2018, 11, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R. Cytomegalovirus Infection after Liver Transplantation: Current Concepts and Challenges. World J. Gastroenterol. 2008, 14, 4849–4860. [Google Scholar] [CrossRef]
- Phan, T.L.; Carlin, K.; Ljungman, P.; Politikos, I.; Boussiotis, V.; Boeckh, M.; Shaffer, M.L.; Zerr, D.M. Human Herpesvirus-6B Reactivation Is a Risk Factor for Grades II to IV Acute Graft-versus-Host Disease after Hematopoietic Stem Cell Transplantation: A Systematic Review and Meta-Analysis. Biol. Blood Marrow Transplant. 2018, 24, 2324–2336. [Google Scholar] [CrossRef]
- Camille, R.; Massih, A.; Razonable, R.R.; Submissions, O.; Com, W.W. Human Herpesvirus 6 Infections after Liver Transplantation. World J. Gastroenterol. 2009, 15, 2561–2569. [Google Scholar] [CrossRef]
- Jakharia, N.; Howard, D.; Riedel, D.J. CMV Infection in Hematopoietic Stem Cell Transplantation: Prevention and Treatment Strategies. Curr. Treat. Options Infect. Dis. 2021, 13, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Manuel, O.; Avery, R.K. Update on Cytomegalovirus in Transplant Recipients: New Agents, Prophylaxis, and Cell-Mediated Immunity. Curr. Opin. Infect. Dis. 2021, 34, 307–313. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 324. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Overwhelming Evidence for a Major Role for Herpes Simplex Virus Type 1 (Hsv1) in Alzheimer’s Disease (Ad); Underwhelming Evidence Against. Vaccines 2021, 9, 679. [Google Scholar] [CrossRef]
- Wozniak, M.; Mee, A.; Itzhaki, R.F. Herpes Simplex Virus Type 1 DNA Is Located within Alzheimer’s Disease Amyloid Plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef]
- Harris, S.A.; Harris, E.A. Molecular Mechanisms for Herpes Simplex Virus Type 1 Pathogenesis in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Marcocci, M.E.; Sgarbanti, R.; Civitelli, L.; Ripoli, C.; Piacentini, R.; Garaci, E.; Grassi, C.; Palamara, A.T. Infectious Agents and Neurodegeneration. Mol. Neurobiol. 2012, 46, 614–638. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limongi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent Herpes Simplex Virus-1 Infection Induces Hallmarks of Neurodegeneration and Cognitive Deficits in Mice. PLoS Pathog. 2019, 15, e1007617. [Google Scholar] [CrossRef]
- Napoletani, G.; Protto, V.; Marcocci, M.E.; Nencioni, L.; Palamara, A.T.; De Chiara, G. Recurrent Herpes Simplex Virus Type 1 (Hsv-1) Infection Modulates Neuronal Aging Marks in in Vitro and in Vivo Models. Int. J. Mol. Sci. 2021, 22, 6279. [Google Scholar] [CrossRef]
- Readhead, B.; Haure-Mirande, J.V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018, 99, 64–82.e7. [Google Scholar] [CrossRef]
- Welling, M.M.; Nabuurs, R.J.A.; van der Weerd, L. Potential Role of Antimicrobial Peptides in the Early Onset of Alzheimer’s Disease. Alzheimer’s Dement. 2015, 11, 51–57. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The Antimicrobial Protection Hypothesis of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated with Multiple Sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Tzeng, N.-S.; Chung, C.-H.; Lin, F.-H.; Chiang, C.-P.; Yeh, C.-B.; Huang, S.-Y.; Lu, R.-B.; Chang, H.-A.; Kao, Y.-C.; Yeh, H.-W.; et al. Anti-Herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections-a Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics 2018, 15, 417–429. [Google Scholar] [CrossRef]
- Linard, M.; Bezin, J.; Hucteau, E.; Joly, P.; Garrigue, I.; Dartigues, J.F.; Pariente, A.; Helmer, C. Antiherpetic Drugs: A Potential Way to Prevent Alzheimer’s Disease? Alzheimer’s Res. Ther. 2022, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.E.; Weller, S.K. The Role of DNA Recombination in Herpes Simplex Virus DNA Replication. IUBMB Life 2003, 55, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Reuven, N.; Mohni, K.; Schumacher, A.; Weller, S.K. Structure of the Herpes Simplex Virus 1 Genome: Manipulation of Nicks and Gaps Can Abrogate Infectivity and Alter the Cellular DNA Damage Response. J. Virol. 2014, 88, 10146–10156. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Weller, S.K. HSV-I and the Cellular DNA Damage Response. Future Virol. 2015, 10, 383–397. [Google Scholar] [CrossRef]
- Shamoo, Y.; Steitz, T.A. Building a Replisome from Interacting Pieces: Sliding Clamp Complexed to a Peptide from Dna Polymerase and a Polymerase Editing Complex. Cell 1999, 99, 155–166. [Google Scholar] [CrossRef]
- Liu, S.; Knafels, J.D.; Chang, J.S.; Waszak, G.A.; Baldwin, E.T.; Deibel, M.R.; Thomsen, D.R.; Homa, F.L.; Wells, P.A.; Tory, M.C.; et al. Crystal Structure of the Herpes Simplex Virus 1 DNA Polymerase. J. Biol. Chem. 2006, 281, 18193–18200. [Google Scholar] [CrossRef]
- Bennett, N.; Götte, M. Utility of the Bacteriophage RB69 Polymerase Gp43 as a Surrogate Enzyme for Herpesvirus Orthologs. Viruses 2013, 5, 54–86. [Google Scholar] [CrossRef]
- Xia, S.; Konigsberg, W.H. RB69 DNA Polymerase Structure, Kinetics, and Fidelity. Biochemistry 2014, 53, 2752–2767. [Google Scholar] [CrossRef]
- Kazlauskas, D.; Krupovic, M.; Guglielmini, J.; Forterre, P.; Venclovas, C.S. Diversity and Evolution of B-Family DNA Polymerases. Nucleic Acids Res. 2020, 48, 10142–10156. [Google Scholar] [CrossRef]
- Hayward, G.S.; Frenkel, N.; Roizman, B. Anatomy of Herpes Simplex Virus DNA: Strain Differences and Heterogeneity in the Locations of Restriction Endonuclease Cleavage Sites. Proc. Natl. Acad. Sci. USA 1975, 72, 1768–1772. [Google Scholar] [CrossRef]
- Kintner, R.L.; Allan, R.W.; Brandt, C.R. Recombinants Are Isolated at High Frequency Following in Vivo Mixed Ocular Infection with Two Avirulent Herpes Simplex Virus Type 1 Strains. Arch. Virol. 1995, 140, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Bowden, R.J.; Sakaoka, H.; Donnelly, P.; Ward, R. High Recombination Rate in Herpes Simplex Virus Type 1 Natural Populations Suggests Significant Co-Infection. Infect. Genet. Evol. 2004, 4, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Lingen, M.; Hengerer, F.; Falke, D. Mixed Vaginal Infections of Balb/c Mice with Low Virulent Herpes Simplex Type 1 Strains Result in Restoration of Virulence Properties: Vaginitis/Vulvitis and Neuroinvasiveness. Med. Microbiol. Immunol. 1997, 185, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Szpara, M.L.; Gatherer, D.; Ochoa, A.; Greenbaum, B.; Dolan, A.; Bowden, R.J.; Enquist, L.W.; Legendre, M.; Davison, A.J. Evolution and Diversity in Human Herpes Simplex Virus Genomes. J. Virol. 2014, 88, 1209–1227. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.J.; Roizman, B.; Kovler, M.B. Anatomy of Herpes Simplex Virus DNA VIII. Properties of the Replicating DNA. J. Virol. 1977, 23, 394–411. [Google Scholar] [CrossRef]
- Severini, A.; Scraba, D.G.; Lorne, A.D.; Tyrrell, J. Branched Structures in the Intracellular DNA of Herpes Simplex Virus Type 1. J. Virol. 1996, 70, 3169–3175. [Google Scholar] [CrossRef]
- Severini, A.; Morgan, A.R.; Tovell, D.R.; Tyrrell, D.L.J. Study of the Structure of Replicative Intermediates of HSV-1 DNA by Pulsed-Field Gel Electrophoresis. Virology 1994, 200, 428–435. [Google Scholar] [CrossRef]
- Martinez, R.; Sarisky, R.T.; Weber, P.; Weller, S.K. Herpes Simplex Virus Type 1 Alkaline Nuclease Is Required for Efficient Processing of Viral DNA Replication Intermediates. J. Virol. 1996, 70, 2075–2085. [Google Scholar] [CrossRef]
- Bacon, T.H.; Levin, M.J.; Leary, J.J.; Sarisky, R.T.; Sutton, D. Herpes Simplex Virus Resistance to Acyclovir and Penciclovir after Two Decades of Antiviral Therapy. Clincal Microbiol. Rev. 2003, 16, 114–128. [Google Scholar] [CrossRef]
- Levin, M.J.; Bacon, T.H.; Leary, J.J. Resistance of Herpes Simplex Virus Infections to Nucleoside Analogues in HIV-Infected Patients. Clin. Infect. Dis. 2004, 39 (Suppl. 5), S248–S257. [Google Scholar] [CrossRef]
- Chen, K.; Cheng, M.P.; Hammond, S.P.; Einsele, H.; Marty, F.M. Antiviral Prophylaxis for Cytomegalovirus Infection in Allogeneic Hematopoietic Cell Transplantation. Blood Adv. 2018, 2, 2159–2175. [Google Scholar] [CrossRef] [PubMed]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir Prophylaxis for Cytomegalovirus in Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef] [PubMed]
- Chou, S. Rapid In Vitro Evolution of Human Cytomegalovirus UL56 Mutations That Confer Letermovir Resistance Downloaded From. Antimicrob. Agents Chemother. 2015, 59, 6588–6593. [Google Scholar] [CrossRef] [PubMed]
- Chou, S. A Third Component of the Human Cytomegalovirus Terminase Complex Is Involved in Letermovir Resistance. Antiviral Res. 2017, 148, 1–4. [Google Scholar] [CrossRef]
- Cherrier, L.; Nasar, A.; Goodlet, K.J.; Nailor, M.D.; Tokman, S.; Chou, S. Emergence of Letermovir Resistance in a Lung Transplant Recipient with Ganciclovir-Resistant Cytomegalovirus Infection. Am. J. Transplant. 2018, 18, 3060–3064. [Google Scholar] [CrossRef]
- Lischka, P.; Michel, D.; Zimmermann, H. Characterization of Cytomegalovirus Breakthrough Events in a Phase 2 Prophylaxis Trial of Letermovir (AIC246, MK 8228). J. Infect. Dis. 2016, 213, 23–30. [Google Scholar] [CrossRef] [PubMed]
- McCrea, J.B.; Macha, S.; Adedoyin, A.; Marshall, W.; Menzel, K.; Cho, C.R.; Liu, F.; Zhao, T.; Levine, V.; Kraft, W.K.; et al. Pharmacokinetic Drug-Drug Interactions Between Letermovir and the Immunosuppressants Cyclosporine, Tacrolimus, Sirolimus, and Mycophenolate Mofetil. J. Clin. Pharmacol. 2019, 59, 1331–1339. [Google Scholar] [CrossRef]
- Frange, P.; Leruez-Ville, M. Maribavir, Brincidofovir and Letermovir: Efficacy and Safety of New Antiviral Drugs for Treating Cytomegalovirus Infections. Médecine Mal. Infect. 2018, 48, 495–502. [Google Scholar] [CrossRef]
- Krosky, P.M.; Baek, M.-C.; Coen, D.M. The Human Cytomegalovirus UL97 Protein Kinase, an Antiviral Drug Target, Is Required at the Stage of Nuclear Egress. J. Virol. 2003, 77, 905–914. [Google Scholar] [CrossRef]
- Wald, A.; Corey, L.; Timmler, B.; Magaret, A.; Warren, T.; Tyring, S.; Johnston, C.; Kriesel, J.; Fife, K.; Galitz, L.; et al. Helicase-Primase Inhibitor Pritelivir for HSV-2 Infection. N. Engl. J. Med. 2014, 370, 201–210. [Google Scholar] [CrossRef]
- Gorbalenya, A.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the Largest RNA Virus Genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavi. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef]
- Holmes, K.V. SARS Coronavirus: A New Challenge for Prevention and Therapy. J. Clin. Investig. 2003, 111, 1605–1609. [Google Scholar] [CrossRef]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef]
- Chen, B.; Tian, E.K.; He, B.; Tian, L.; Han, R.; Wang, S.; Xiang, Q.; Zhang, S.; El Arnaout, T.; Cheng, W. Overview of Lethal Human Coronaviruses. Signal. Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef]
- Snijder, E.J.; Bredenbeek, P.J.; Dobbe, J.C.; Thiel, V.; Ziebuhr, J.; Poon, L.L.M.; Guan, Y.; Rozanov, M.; Spaan, W.J.M.; Gorbalenya, A.E. Unique and Conserved Features of Genome and Proteome of SARS-Coronavirus, an Early Split-off from the Coronavirus Group 2 Lineage. J. Mol. Biol. 2003, 331, 991–1004. [Google Scholar] [CrossRef]
- De Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.M.; Galiano, M.; Gorbalenya, A.; Memish, Z.A.; et al. Commentary: Middle East Respiratory Syndrome Coronavirus (MERS-CoV): Announcement of the Coronavirus Study Group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Ahmed, R.; Hasan, R.; Siddiki, A.M.A.M.Z.; Islam, M.S. Host Range Projection of SARS-CoV-2: South Asia Perspective. Infect. Genet. Evol. 2021, 87, 104670. [Google Scholar] [CrossRef]
- Do Vale, B.; Lopes, A.P.; Fontes, M.d.C.; Silvestre, M.; Cardoso, L.; Coelho, A.C. Bats, Pangolins, Minks and Other Animals - Villains or Victims of SARS-CoV-2? Vet. Res. Commun. 2021, 45, 1–19. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Luk, H.K.H.; Wong, A.C.P.; Li, K.S.M.; Zhu, L.; He, Z.; Fung, J.; Chan, T.T.Y.; Fung, K.S.C.; Woo, P.C.Y. Possible Bat Origin of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. J. 2020, 26, 1542. [Google Scholar] [CrossRef]
- Singh, J.; Pandit, P.; McArthur, A.G.; Banerjee, A.; Mossman, K.L. Evolutionary Trajectory of SARS-CoV-2 and Emerging Variants. Virol. J. 2021, 18, 1–21. [Google Scholar] [CrossRef]
- Becker, D.J.; Albery, G.F.; Sjodin, A.R.; Poisot, T.; Bergner, L.M.; Chen, B.; Cohen, L.E.; Dallas, T.A.; Eskew, E.A.; Fagre, A.C.; et al. Optimising Predictive Models to Prioritise Viral Discovery in Zoonotic Reservoirs. Lancet Microbe 2022, 5247, 1–13. [Google Scholar] [CrossRef]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The Coronavirus Proofreading Exoribonuclease Mediates Extensive Viral Recombination. PLoS Pathog. 2021, 17, 1–28. [Google Scholar] [CrossRef]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA Virus 3′→5′ Exoribonuclease That Is Critically Involved in Coronavirus RNA Synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High Fidelity of Murine Hepatitis Virus Replication Is Decreased in Nsp14 Exoribonuclease Mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Becker, M.M.; Halpin, R.A.; Li, K.; Venter, E.; Lu, X.; Scherbakova, S.; Graham, R.L.; Baric, R.S.; Stockwell, T.B.; et al. Infidelity of SARS-CoV Nsp14-Exonuclease Mutant Virus Replication Is Revealed by Complete Genome Sequencing. PLoS Pathog. 2010, 6, e1000896. [Google Scholar] [CrossRef]
- Denison, M.R.; Graham, R.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA Proofreading Machine Regulates Replication Fidelity and Diversity. RNA Biol. 2011, 8, 270–289. [Google Scholar] [CrossRef]
- Smith, E.C.; Blanc, H.; Vignuzzi, M.; Denison, M.R. Coronaviruses Lacking Exoribonuclease Activity Are Susceptible to Lethal Mutagenesis: Evidence for Proofreading and Potential Therapeutics. PLoS Pathog. 2013, 9, e1003565. [Google Scholar] [CrossRef]
- Smith, E.C.; Case, J.B.; Blanc, H.; Isakov, O.; Shomron, N.; Vignuzzi, M.; Denison, M.R. Mutations in Coronavirus Nonstructural Protein 10 Decrease Virus Replication Fidelity. J. Virol. 2015, 89, 6418–6426. [Google Scholar] [CrossRef] [PubMed]
- Case, J.B.; Li, Y.; Elliott, R.; Lu, X.; Graepel, K.W.; Sexton, N.R.; Smith, E.C.; Weiss, S.R.; Denison, M.R. Murine Hepatitis Virus Nsp14 Exoribonuclease Activity Is Required for Resistance to Innate Immunity. J. Virol. 2018, 92, e01531-17. [Google Scholar] [CrossRef] [PubMed]
- Graepel, K.W.; Agostini, M.L.; Lu, X.; Sexton, N.R.; Denison, M.R. Fitness Barriers Limit Reversion of a Proofreading-Deficient Coronavirus. J. Virol. 2019, 93, e00711-19. [Google Scholar] [CrossRef] [PubMed]
- Becares, M.; Pascual-iglesias, A.; Nogales, A.; Sola, I.; Enjuanes, L.; Zuñiga, S. Mutagenesis of Coronavirus Nsp14 Reveals Its Potential Role In. J. Virol. 2016, 90, 5399–5414. [Google Scholar] [CrossRef] [PubMed]
- Hirst, G. Genetic Recombination with Newcastle Disease Virus, Polioviruses, and Influenza. In Cold Spring Harbor symposia on quantitative biology; Cold Spring Harbor: Long Island, NY, USA, 1962; Volume 27, pp. 303–309. [Google Scholar]
- Ledinko, N. Genetic Recombination with Poliovirus Type 1. Virology 1963, 20, 107–119. [Google Scholar] [CrossRef]
- Cooper, P.D. Genetics of Picornaviruses. In Regulation and Genetics; Franenkel-Conrat, H., Ed.; Springer: Boston, MA USA, 1977; pp. 133–207. [Google Scholar] [CrossRef]
- Kirkegaard, K.; Baltimore, D. The Mechanism of RNA Recombination in Poliovirus. Cell 1986, 47, 433–443. [Google Scholar] [CrossRef]
- Lai, M.M.; Baric, R.S.; Makino, S.; Keck, J.G.; Egbert, J.; Leibowitz, J.L.; Stohlman, S.A. Recombination between Nonsegmented RNA Genomes of Murine Coronaviruses. J. Virol. 1985, 56, 449–456. [Google Scholar] [CrossRef]
- Makino, S.; Keck, J.G.; Stohlman, S.A.; Lai, M.M. High-Frequency RNA Recombination of Murine Coronaviruses. J. Virol. 1986, 57, 729–737. [Google Scholar] [CrossRef]
- Keck, J.G.; Matsushima, G.K.; Makino, S.; Fleming, J.O.; Vannier, D.M.; Stohlman, S.A.; Lai, M.M. In Vivo RNA-RNA Recombination of Coronavirus in Mouse Brain. J. Virol. 1988, 62, 1810–1813. [Google Scholar] [CrossRef]
- Rowe, C.L.; Baker, S.C.; Nathan, M.J.; Sgro, J.Y.; Palmenberg, A.C.; Fleming, J.O. Quasispecies Development by High Frequency RNA Recombination during MHV Persistence. Adv. Exp. Med. Biol. 1998, 440, 759–765. [Google Scholar] [CrossRef]
- Chen, N.; Li, S.; Zhou, R.; Zhu, M.; He, S.; Ye, M.; Huang, Y.; Li, S.; Zhu, C.; Xia, P.; et al. Two Novel Porcine Epidemic Diarrhea Virus (PEDV) Recombinants from a Natural Recombinant and Distinct Subtypes of PEDV Variants. Virus Res. 2017, 242, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.Y.; Chen, T.; Zhang, X.; Shao, G.M.; Cao, Y.; Chen, D.K.; Lin, W.C.; Chen, F.; Xie, Q.M. Molecular Characteristic and Pathogenicity Analysis of a Virulent Recombinant Avain Infectious Bronchitis Virus Isolated in China. Poult. Sci. 2018, 97, 3519–3531. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Dinnon, K.H.; Yount, B.L.; McAnarney, E.T.; Gralinski, L.E.; Hale, A.; Graham, R.L.; Scobey, T.; Anthony, S.J.; Wang, L.; et al. Trypsin Treatment Unlocks Barrier for Zoonotic Bat Coronavirus Infection. J. Virol. 2019, 94, e01774-19. [Google Scholar] [CrossRef] [PubMed]
- Domingo, J.L. What We Know and What We Need to Know about the Origin of SARS-CoV-2. Environ. Res. 2021, 200, 111785. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Yi, H. 2019 Novel Coronavirus Is Undergoing Active Recombination. Clin. Infect. Dis. 2020, 71, 884–885. [Google Scholar] [CrossRef]
- Ziebuhr, J. The Coronavirus Replicase. In Coronavirus Replication and Reverse Genetics; Enjuanes, L., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 57–94. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A. Virus-Encoded Proteinases and Proteolytic Processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef]
- Sawicki, S.G.; Sawicki, D.L.; Younker, D.; Meyer, Y.; Thiel, V.; Stokes, H.; Siddell, S.G. Functional and Genetic Analysis of Coronavirus Replicase-Transcriptase Proteins. PLoS Pathog. 2005, 1, 0310–0322. [Google Scholar] [CrossRef]
- Sawicki, S.G.; Sawicki, D.L.; Siddell, S.G. A Contemporary View of Coronavirus Transcription. J. Virol. 2007, 81, 20–29. [Google Scholar] [CrossRef]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef]
- Snijder, E.J.; Limpens, R.W.A.L.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.F.G.A.; Koster, A.J.; Bárcena, M. A Unifying Structural and Functional Model of the Coronavirus Replication Organelle: Tracking down RNA Synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses: Methods and Protocols; Maier, H.J., Bickerton, E., Britton, P., Eds.; Springer: New York, NY, USA, 2015; pp. 1–23. [Google Scholar] [CrossRef]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016, 96, 59–126. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, A.A.; Gorbalenya, A. A Nidovirus Perspective on SARS-CoV-2. Biochem. Biophys. Res. Commun. 2021, 538, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Grellet, E.; L’Hôte, I.; Goulet, A.; Imbert, I. Replication of the Coronavirus Genome: A Paradox among Positive-Strand RNA Viruses. J. Biol. Chem. 2022, 298, 101923. [Google Scholar] [CrossRef]
- Elena, S.F.; Carrasco, P.; Daròs, J.A.; Sanjuán, R. Mechanisms of Genetic Robustness in RNA Viruses. EMBO Rep. 2006, 7, 168–173. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV Nsp12 Polymerase Bound to Nsp7 and Nsp8 Co-Factors. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Zhai, Y.; Sun, F.; Li, X.; Pang, H.; Xu, X.; Bartlam, M.; Rao, Z. Insights into SARS-CoV Transcription and Replication from the Structure of the Nsp7-Nsp8 Hexadecamer. Nat. Struct. Mol. Biol. 2005, 12, 980–986. [Google Scholar] [CrossRef]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One Severe Acute Respiratory Syndrome Coronavirus Protein Complex Integrates Processive RNA Polymerase and Exonuclease Activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef]
- Ferron, F.; Subissi, L.; De Morais, A.T.S.; Le, N.T.T.; Sevajol, M.; Gluais, L.; Decroly, E.; Vonrhein, C.; Bricogne, G.; Canard, B.; et al. Structural and Molecular Basis of Mismatch Correction and Ribavirin Excision from Coronavirus RNA. Proc. Natl. Acad. Sci. USA 2018, 115, E162–E171. [Google Scholar] [CrossRef]
- Yan, L.; Yang, Y.; Li, M.; Zhang, Y.; Zheng, L.; Ge, J.; Huang, Y.C.; Liu, Z.; Wang, T.; Gao, S.; et al. Coupling of N7-Methyltransferase and 3′-5′ Exoribonuclease with SARS-CoV-2 Polymerase Reveals Mechanisms for Capping and Proofreading. Cell 2021, 184, 3474–3485.e11. [Google Scholar] [CrossRef]
- Bouvet, M.; Imbert, I.; Subissi, L.; Gluais, L.; Canard, B.; Decroly, E. RNA 3′-End Mismatch Excision by the Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein Nsp10/Nsp14 Exoribonuclease Complex. Proc. Natl. Acad. Sci. USA 2012, 109, 9372–9377. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural Basis and Functional Analysis of the SARS Coronavirus Nsp14-Nsp10 Complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef]
- Lin, S.; Chen, H.; Chen, Z.; Yang, F.; Ye, F.; Zheng, Y.; Yang, J.; Lin, X.; Sun, H.; Wang, L.; et al. Crystal Structure of SARS-CoV-2 Nsp10 Bound to Nsp14-ExoN Domain Reveals an Exoribonuclease with Both Structural and Functional Integrity. Nucleic Acids Res. 2021, 49, 5382–5392. [Google Scholar] [CrossRef]
- Deval, J.; Gurard-levin, Z.A. Opportunities and Challenges in Targeting the Proofreading Activity of SARS-CoV-2 Polymerase Complex. Molecules 2022, 27, 2918. [Google Scholar] [CrossRef] [PubMed]
- Ogando, N.S.; Ferron, F.; Decroly, E.; Canard, B.; Posthuma, C.C.; Snijder, E.J. The Curious Case of the Nidovirus Exoribonuclease: Its Role in RNA Synthesis and Replication Fidelity. Front. Microbiol. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Smith, E.C.; Denison, M.R. Coronaviruses as DNA Wannabes: A New Model for the Regulation of RNA Virus Replication Fidelity. PLoS Pathog. 2013, 9, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Schooley, R.T.; Carlin, A.F.; Beadle, J.R.; Valiaeva, N.; Zhang, X.-Q.; Clark, A.E.; McMillan, R.E.; Leibel, S.L.; McVicar, R.N.; Xie, J.; et al. Rethinking Remdesivir: Synthesis, Antiviral Activity, and Pharmacokinetics of Oral Lipid Prodrugs. Antimicrob. Agents Chemother. 2021, 65, e01155-21. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.A.; Eron, J.J.; Holman, W.; Cohen, M.S.; Fang, L.; Szewczyk, L.J.; Sheahan, T.P.; Baric, R.; Mollan, K.R.; Wolfe, C.R.; et al. A Phase 2a Clinical Trial of Molnupiravir in Patients with COVID-19 Shows Accelerated SARS-CoV-2 RNA Clearance and Elimination of Infectious Virus. Sci. Transl. Med. 2022, 14, 1–15. [Google Scholar] [CrossRef]
- Rubin, R. From Positive to Negative to Positive Again—The Mystery of Why COVID-19 Rebounds in Some Patients Who Take Paxlovid. JAMA 2022, 327, 2380–2382. [Google Scholar] [CrossRef]
- Okano, K.; Vanarsdall, A.L.; Rohrmann, G.F. Characterization of a Baculovirus Lacking the Alkaline Nuclease Gene. J. Virol. 2004, 78, 10650–10656. [Google Scholar] [CrossRef]
- Gammon, D.B.; Evans, D.H. The 3′-to-5′ Exonuclease Activity of Vaccinia Virus DNA Polymerase Is Essential and Plays a Role in Promoting Virus Genetic Recombination. J. Virol. 2009, 83, 4236–4250. [Google Scholar] [CrossRef] [PubMed]
- Vallée, G.; Norris, P.; Paszkowski, P.; Noyce, R.S.; Evans, D.H. Vaccinia Virus Gene Acquisition through Nonhomologous Recombination. J. Virol. 2021, 95, e00318-21. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. Nucleases: Diversity of Structure, Function and Mechanism. Q. Rev. Biophys. 2011, 44, 1–93. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.A.; Johnson, A.A.; Marchand, C.; Davis, D.A.; Yarchoan, R.; Pommier, Y. Preferential Inhibition of the Magnesium-Dependent Strand Transfer Reaction of HIV-1 Integrase by -Hydroxytropolones. Mol. Pharmacol. 2006, 69, 1454–1460. [Google Scholar] [CrossRef]
- Li, Y.; Xuan, S.; Feng, Y.; Yan, A. Targeting HIV-1 Integrase with Strand Transfer Inhibitors. Drug Discov. Today 2015, 20, 435–449. [Google Scholar] [CrossRef]
- Esposito, F.; Tramontano, E. Past and Future. Current Drugs Targeting HIV-1 Integrase and Reverse Transcriptase-Associated Ribonuclease H Activity: Single and Dual Active Site Inhibitors. Antivir. Chem. Chemother. 2014, 23, 129–144. [Google Scholar] [CrossRef]
- Stevaert, A.; Naesens, L. The Influenza Virus Polymerase Complex: An Update on Its Structure, Functions, and Significance for Antiviral Drug Design. Med. Res. Rev. 2016, 36, 1127–1173. [Google Scholar] [CrossRef]
- Bujnicki, J.M.; Rychlewski, L. Grouping Together Highly Diverged PD-(D/E)XK Nucleases and Identification of Novel Superfamily Members Using Structure-Guided Alignment of Sequence Profiles. J. Mol. Microbiol. Biotechnol. 2001, 3, 69–72. [Google Scholar]
- Sigamani, S.S.; Zhao, H.; Kamau, Y.N.; Baines, J.D.; Tang, L. The Structure of the Herpes Simplex Virus DNA-Packaging Terminase PUL15 Nuclease Domain Suggests an Evolutionary Lineage among Eukaryotic and Prokaryotic Viruses. J. Virol. 2013, 87, 7140–7148. [Google Scholar] [CrossRef]
- Majorek, K.A.; Dunin-Horkawicz, S.; Steczkiewicz, K.; Muszewska, A.; Nowotny, M.; Ginalski, K.; Bujnicki, J.M. The RNase H-like Superfamily: New Members, Comparative Structural Analysis and Evolutionary Classification. Nucleic Acids Res. 2014, 42, 4160–4179. [Google Scholar] [CrossRef]
- Baylis, S.A.; Purifoyl, D.J.M.; Littler, E. The Characterization of the EBV Alkaline Deoxyribonuclease Cloned and Expressed in E.Coli. Nucleic Acids Res. 1989, 17, 7609–7622. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.F.; Hsu, T.Y.; Liu, M.Y.; Lin, L.S.; Yang, H.L.; Chen, J.Y.; Yang, C.S. Characterization of Epstein-Barr Virus DNase and Its Interaction with the Major DNA Binding Protein. Virology 1995, 208, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Stolzenberg, M.-C.; Ooka, T. Purification and Properties of Epstein-Barr Virus DNase Expressed in Escherichia Coli. J. Virol. 1990, 64, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Sheaffer, A.K.; Weinheimer, S.P.; Tenney, D.J. The Human Cytomegalovirus UL98 Gene Encodes the Conserved Herpesvirus Alkaline Nuclease. J. Gen. Virol. 1997, 78, 2953–2961. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goldstein, J.; Weller, S. The Exonuclease Activity of HSV-1 UL12 Is Required for in Vivo Function. Virology 1998, 244, 442–457. [Google Scholar] [CrossRef]
- Shao, L.; Rapp, L.; Weller, S.K. Herpes Simplex Virus 1 Alkaline Nuclease Is Required for Efficient Egress of Capsids from the Nucleus. Virology 1993, 196, 146–162. [Google Scholar] [CrossRef]
- Gao, M.; Robertson, B.J.; McCann III, P.J.; O’Boyle II, D.R.; Weller, S.K.; Newcomb, W.W.; Brown, J.C.; Weinheimer, S.P. Functional Conservations of the Alkaline Nuclease of Herpes Simplex Type 1 and Human Cytomegalovirus. Virology 1998, 249, 460–470. [Google Scholar] [CrossRef][Green Version]
- Yu, D.; Silva, M.C.; Shenk, T. Functional Map of Human Cytomegalovirus AD169 Defined by Global Mutational Analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 12396–12401. [Google Scholar] [CrossRef]
- Feederle, R.; Bannert, H.; Lips, H.; Müller-Lantzsch, N.; Delecluse, H.-J. The Epstein-Barr Virus Alkaline Exonuclease BGLF5 Serves Pleiotropic Functions in Virus Replication. J. Virol. 2009, 83, 4952–4962. [Google Scholar] [CrossRef]
- Duguay, B.A.; Saffran, H.A.; Ponomarev, A.; Duley, S.A.; Eaton, H.E.; Smiley, J.R.; Sandri-Goldin, R.M. Elimination of Mitochondrial DNA Is Not Required for Herpes Simplex Virus 1 Replication Brett. J. Virol. 2014, 88, 2967–2976. [Google Scholar] [CrossRef]
- Buisson, M.; Géoui, T.; Flot, D.; Tarbouriech, N.; Ressing, M.E.; Wiertz, E.J.; Burmeister, W.P. A Bridge Crosses the Active-Site Canyon of the Epstein-Barr Virus Nuclease with DNase and RNase Activities. J. Mol. Biol. 2009, 391, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Burmeister, W.P.; Boer, I.G.J.; Van Leeuwen, D.; Buisson, M.; Gorbalenya, A.; Wiertz, E.J.H.J.; Ressing, M.E. The “Bridge” in the Epstein-Barr Virus Alkaline Exonuclease Protein BGLF5 Contributes to Shutoff Activity during Productive Infection. J. Virol. 2012, 86, 9175–9187. [Google Scholar] [CrossRef] [PubMed]
- Bagnéris, C.; Briggs, L.C.; Savva, R.; Ebrahimi, B.; Barrett, T.E. Crystal Structure of a KSHV-SOX-DNA Complex: Insights into the Molecular Mechanisms Underlying DNase Activity and Host Shutoff. Nucleic Acids Res. 2011, 39, 5744–5756. [Google Scholar] [CrossRef] [PubMed]
- Dahlroth, S.-L.L.; Gurmu, D.; Haas, J.; Erlandsen, H.; Nordlund, P.R. Crystal Structure of the Shutoff and Exonuclease Protein from the Oncogenic Kaposi’s Sarcoma-Associated Herpesvirus. FEBS J. 2009, 276, 6636–6645. [Google Scholar] [CrossRef]
- Elion, G.B. Mechanism of Action and Selectivity of Acyclovir. Am. J. Med. 1982, 73, 7–13. [Google Scholar] [CrossRef]
- Piret, J.; Boivin, G. Resistance of Herpes Simplex Viruses to Nucleoside Analogues: Mechanisms, Prevalence, and Management. Antimicrob. Agents Chemother. 2011, 55, 459–472. [Google Scholar] [CrossRef]
- Whitley, R.; Baines, J.D. Clinical Management of Herpes Simplex Virus Infections: Past, Present, and Future [Version 1; Peer Review: 2 Approved]. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- James, S.H.; Prichard, M.; Mené Ndez-Arias, L.; Richman, D.D. Current and Future Therapies for Herpes Simplex Virus Infections: Mechanism of Action and Drug Resistance. Curr. Opin. Virol. 2014, 8, 54–61. [Google Scholar] [CrossRef]
- Hwang, Y.T.; Smith, J.F.; Gao, L.; Hwang, C.B. Mutations in the Exo III Motif of the Herpes Simplex Virus DNA Polymerase Gene Can Confer Altered Drug Sensitivities. Virology 1998, 246, 298–305. [Google Scholar] [CrossRef]
- Hwang, Y.T.; Liu, B.-Y.; Hong, C.-Y.; Shillitoe, E.J.; Hwang, C.B. Effects of Exonuclease Activity and Nucleotide Selectivity of the Herpes Simplex Virus DNA Polymerase on the Fidelity of DNA Replication In Vivo. J. Virol. 1999, 73, 5326–5332. [Google Scholar] [CrossRef]
- Hwang, Y.T.; Hwang, C.B. Exonuclease-Deficient Polymerase Mutant of Herpes Simplex Virus Type 1 Induces Altered Spectra of Mutations. J. Virol. 2003, 77, 2946–2955. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coen, D.M.; Lawler, J.L.; Abraham, J. Herpesvirus DNA Polymerase: Structures, Functions, and Mechanisms. In Enzymes; Elsevier Inc.: Amsterdam, The Netherlands, 2021; Volume 50, pp. 133–178. [Google Scholar] [CrossRef]
- Hsiang, C.Y.; Ho, T.Y. Emodin Is a Novel Alkaline Nuclease Inhibitor That Suppresses Herpes Simplex Virus Type 1 Yields in Cell Cultures. Br. J. Pharmacol. 2008, 155, 227–235. [Google Scholar] [CrossRef]
- Alam, Z.; Al-Mahdi, Z.; Zhu, Y.; Mckee, Z.; Parris, D.S.; Parikh, H.I.; Kellogg, G.E.; Kuchta, A.L.; McVoy, M.A. Anti-Cytomegalovirus Activity of the Anthraquinone Atanyl Blue PRL. Antiviral Res. 2015, 114, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, J.C.; Weber, P.C. A Colorimetric Assay for High-Throughput Screening of Inhibitors of Herpes Simplex Virus Type 1 Alkaline Nuclease. Anal. Biochem. 2001, 293, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.; Weller, S.K. In Vitro Processing of Herpes Simplex Virus Type 1 DNA Replication Intermediates by the Viral Alkaline Nuclease, UL12. J. Virol. 1998, 72, 8772–8781. [Google Scholar] [CrossRef]
- Ireland, P.J.; Tavis, J.; D’Erasmo, M.P.; Hirsch, D.R.; Murelli, R.P.; Cadiz, M.M.; Patel, B.S.; Gupta, A.K.; Edwards, T.C.; Korom, M.; et al. Synthetic α-Hydroxytropolones Inhibit Replication of Wild-Type and Acyclovir-Resistant Herpes Simplex Viruses. Antimicrob. Agents Chemother. 2016, 60, 2140–2149. [Google Scholar] [CrossRef]
- Tavis, J.; Wang, H.; Tollefson, A.E.; Ying, B.; Korom, M.; Cheng, X.; Cao, F.; Davis, K.L.; Wold, W.S.M.; Morrison, L.A. Inhibitors of Nucleotidyltransferase Superfamily Enzymes Suppress Herpes Simplex Virus Replication. Antimicrob. Agents Chemother. 2014, 58, 7451–7461. [Google Scholar] [CrossRef]
- DiScipio, K.A.; Weerasooriya, S.; Szczepaniak, R.; Hazeen, A.; Wright, L.R.; Wright, D.L.; Weller, S.K. Two-Metal Ion-Dependent Enzymes as Potential Antiviral Targets in Human Herpesviruses Running. MBio 2022, 13, e03226-21. [Google Scholar] [CrossRef]
- Zuo, Y.; Deutscher, M. Exoribonuclease Superfamilies: Structural Analysis and Phylogenetic Distribution. Nucleic Acids Res. 2001, 29, 1017–1026. [Google Scholar] [CrossRef]
- Canal, B.; Fujisawa, R.; McClure, A.W.; Deegan, T.D.; Wu, M.; Ulferts, R.; Weissmann, F.; Drury, L.S.; Bertolin, A.P.; Zeng, J.; et al. Identifying SARS-CoV-2 Antiviral Compounds by Screening for Small Molecule Inhibitors of Nsp15 Endoribonuclease. Biochem. J. 2021, 478, 2465–2479. [Google Scholar] [CrossRef]
- Tun, M.M.N.; Morita, K.; Ishikawa, T.; Urata, S. The Antiviral Effect of the Chemical Compounds Targeting Ded/Edh Motifs of the Viral Proteins on Lymphocytic Choriomeningitis Virus and Sars-Cov-2. Viruses 2021, 13, 1220. [Google Scholar] [CrossRef]
- Rona, G.; Zeke, A.; Miwatani-Minter, B.; de Vries, M.; Kaur, R.; Schinlever, A.; Garcia, S.F.; Goldberg, H.V.; Wang, H.; Hinds, T.R.; et al. The NSP14/NSP10 RNA Repair Complex as a Pan-Coronavirus Therapeutic Target. Cell Death Differ. 2022, 29, 285–292. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, L.R.; Wright, D.L.; Weller, S.K. Viral Nucleases from Herpesviruses and Coronavirus in Recombination and Proofreading: Potential Targets for Antiviral Drug Discovery. Viruses 2022, 14, 1557. https://doi.org/10.3390/v14071557
Wright LR, Wright DL, Weller SK. Viral Nucleases from Herpesviruses and Coronavirus in Recombination and Proofreading: Potential Targets for Antiviral Drug Discovery. Viruses. 2022; 14(7):1557. https://doi.org/10.3390/v14071557
Chicago/Turabian StyleWright, Lee R., Dennis L. Wright, and Sandra K. Weller. 2022. "Viral Nucleases from Herpesviruses and Coronavirus in Recombination and Proofreading: Potential Targets for Antiviral Drug Discovery" Viruses 14, no. 7: 1557. https://doi.org/10.3390/v14071557
APA StyleWright, L. R., Wright, D. L., & Weller, S. K. (2022). Viral Nucleases from Herpesviruses and Coronavirus in Recombination and Proofreading: Potential Targets for Antiviral Drug Discovery. Viruses, 14(7), 1557. https://doi.org/10.3390/v14071557