
Implementation of GA-VirReport, a Web-Based Bioinformatics Toolkit for Post-Entry Quarantine Screening of Virus and Viroids in Plants

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.1.1. Control Plants
2.1.2. Imported Plants
2.2. Nucleic Acid Extraction
2.3. Library Construction and Sequencing
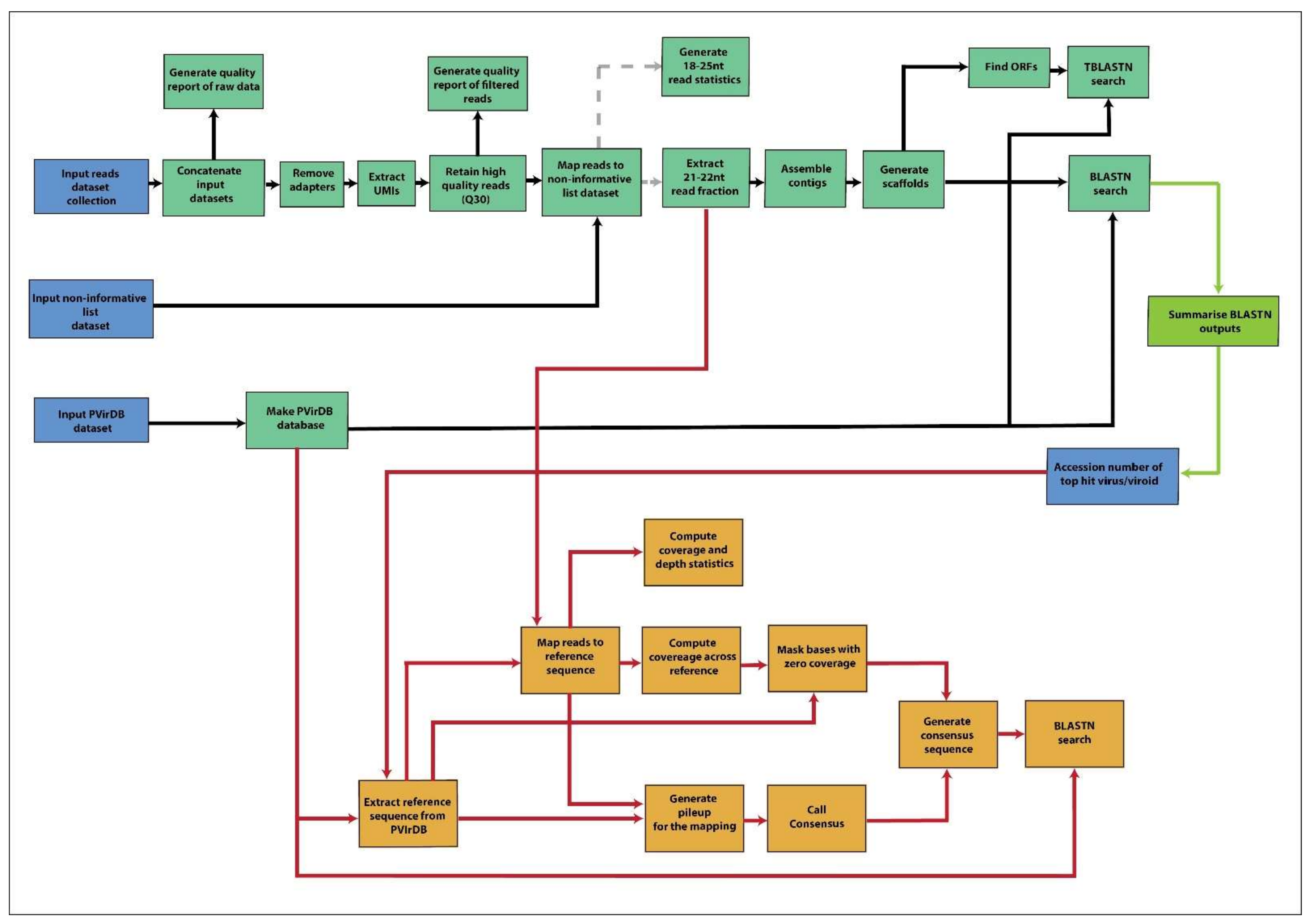
2.4. Bioinformatics Analyses
2.4.1. Preparation of Input Files for the GA-VirReport Workflow
2.4.2. GA-VirReport Workflow
2.4.3. GA-VirReport-Stats Workflow
2.4.4. Curated Plant Virus–Viroid Database: PVirDB
3. Results
3.1. Pilot Study Implementing and Testing the GA-VirReport Bioinformatics Workflow
3.1.1. Sequencing and Assembly Statistics
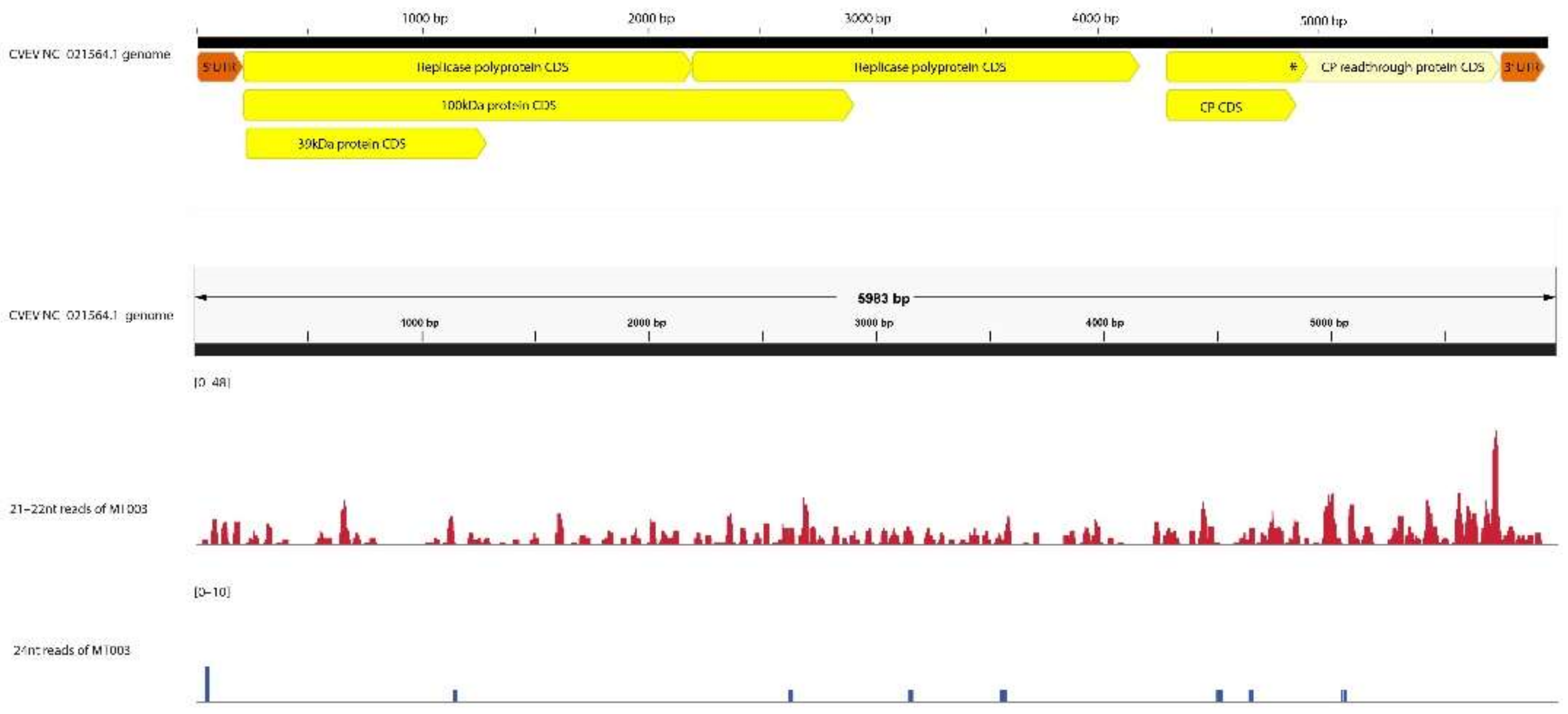
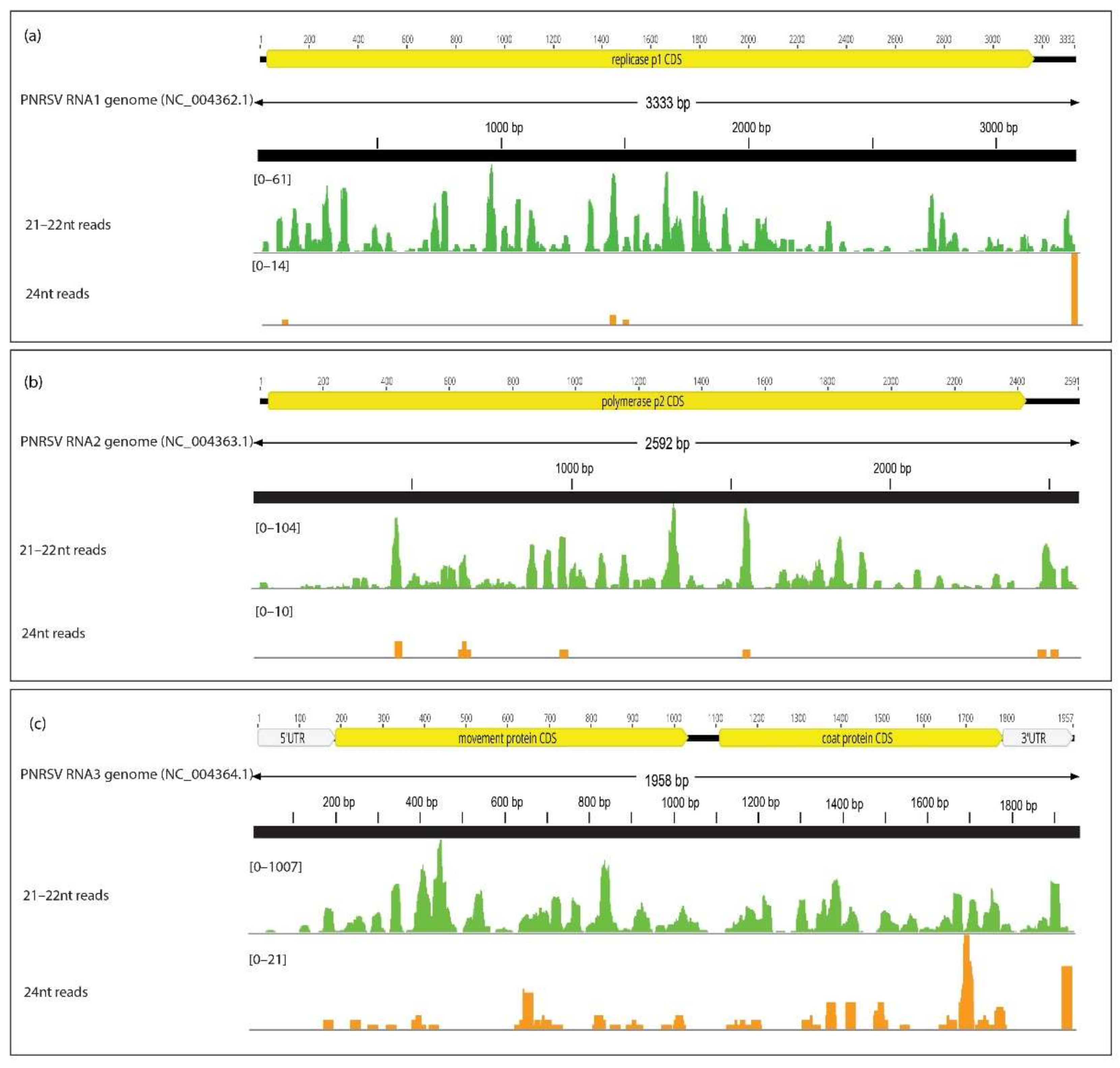
3.1.2. GA-VirReport Detected All PEQ-positive Viruses in Control Plants Using the 21–22 nt Read Fraction
3.1.3. The Sparser Number of Reads Recovered in the 24 nt Read Fraction Can Impair Viral Detection Relying on De Novo Assembly
3.1.4. GA-VirReport Detected Additional Viruses in Control Plants
3.1.5. Cross-Sample Contamination Is Not Detected
3.1.6. Short Contigs of Plant Material Result in Misassignment in Sequence Similarity Searches against the PVirDB
3.1.7. GA-VirReport-Stats Pipeline Produces Longer Consensus Sequences
3.2. Large-Scale Testing and Implementation of GA-VirReport Bioinformatics Workflow on Imported Plants under Quarantine
3.2.1. Sequencing Statistics
3.2.2. GA-VirReport Detected a Variety of Pests in Plants Quarantined at PEQ
3.3. Curation of a Custom Plant Virus and Viroid Blast Database (PVirDB)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, S.; Krainer, K.M.C. Pandemics of People and Plants: Which Is the Greater Threat to Food Security? Mol. Plant 2020, 13, 933–934. [Google Scholar] [CrossRef] [PubMed]
- Rodoni, B. The role of plant biosecurity in preventing and controlling emerging plant virus disease epidemics. Virus Res. 2009, 141, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Whattam, M.; Dinsdale, A.; Elliott, C. Evolution of Plant Virus Diagnostics Used in Australian Post Entry Quarantine. Plants 2021, 10, 1430. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.R.; Constable, F.; Tzanetakis, I.E. Quarantine regulations and the impact of modern detection methods. Ann. Rev. Phytopathol. 2016, 54, 189–205. [Google Scholar] [CrossRef] [Green Version]
- FAO. Framework for pest risk analysis. Int. Stand. Phytosanitary Meas. 2007, 1, 13–14. [Google Scholar]
- Gucek, T.; Trdan, S.; Jakse, J.; Javornik, B.; Matousek, J.; Radisek, S. Diagnostic techniques for viroids. Plant Pathol. 2017, 66, 339–358. [Google Scholar] [CrossRef]
- Varma, A.; Singh, M.K. Chapter 6-Diagnosis of plant virus diseases. In Applied Plant Virology; Awasthi, L.P., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 79–92. [Google Scholar]
- Massart, S.; Olmos, A.; Jijakli, H.; Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014, 188, 90–96. [Google Scholar] [CrossRef]
- Maree, H.J.; Fox, A.; Al Rwahnih, M.; Boonham, N.; Candresse, T. Application of HTS for Routine Plant Virus Diagnostics: State of the Art and Challenges. Front. Plant Sci. 2018, 9, 1082. [Google Scholar] [CrossRef] [Green Version]
- Barrero, R.A.; Napier, K.R.; Cunnington, J.; Liefting, L.; Keenan, S.; Frampton, R.A.; Szabó, T.O.; Bulman, S.; Hunter, A.; Ward, L.; et al. An internet-based bioinformatics toolkit for plant biosecurity diagnosis and surveillance of viruses and viroids. BMC Bioinform. 2017, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Rubio, L.; Galipienso, L.; Ferriol, I. Detection of Plant Viruses and Disease Management: Relevance of Genetic Diversity and Evolution. Front. Plant Sci. 2020, 11, 1092. [Google Scholar] [CrossRef]
- Jones, S.; Baizan-Edge, A.; MacFarlane, S.; Torrance, L. Viral Diagnostics in Plants Using Next Generation Sequencing: Computational Analysis in Practice. Front. Plant Sci. 2017, 8, 1770. [Google Scholar] [CrossRef] [PubMed]
- Bester, R.; Cook, G.; Breytenbach, J.H.J.; Steyn, C.; De Bruyn, R.; Maree, H.J. Towards the validation of high-throughput sequencing (HTS) for routine plant virus diagnostics: Measurement of variation linked to HTS detection of citrus viruses and viroids. Virol. J. 2021, 18, 61. [Google Scholar] [CrossRef] [PubMed]
- Vivek, A.; Zahra, S.; Kumar, S. From current knowledge to best practice: A primer on viral diagnostics using deep sequencing of virus-derived small interfering RNAs (vsiRNAs) in infected plants. Methods 2020, 183, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Velasco, L.; Padilla, C.V. High-Throughput Sequencing of Small RNAs for the Sanitary Certification of Viruses in Grapevine. Front. Plant Sci. 2021, 12, 682879. [Google Scholar] [CrossRef] [PubMed]
- Santala, J.; Valkonen, J.P.T. Sensitivity of Small RNA-Based Detection of Plant Viruses. Front. Microbiol. 2018, 9, 939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massart, S.; Chiumenti, M.; De Jonghe, K.; Glover, R.; Haegeman, A.; Koloniuk, I.; Komínek, P.; Kreuze, J.; Kutnjak, D.; Lotos, L.; et al. Virus Detection by High-Throughput Sequencing of Small RNAs: Large-Scale Performance Testing of Sequence Analysis Strategies. Phytopathology 2019, 109, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.-S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef]
- Ho, T.; Tzanetakis, I.E. Development of a virus detection and discovery pipeline using next generation sequencing. Virology 2014, 471–473, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, M.-E.A.; Lelwala, R.V.; Elliott, C.E.; Windell, C.; Fiorito, S.; Dinsdale, A.; Whattam, M.; Pattemore, J.; Barrero, R.A. Side-by-Side Comparison of Post-Entry Quarantine and High Throughput Sequencing Methods for Virus and Viroid Diagnosis. Biology 2022, 11, 263. [Google Scholar] [CrossRef]
- Qiagen CLC genomics Workbench. Available online: https://digitalinsights.qiagen.com (accessed on 1 July 2022).
- Geneious. Available online: https://www.geneious.com/ (accessed on 8 April 2022).
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- bcl2fastq2 Conversion Software v2.20. Available online: https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html (accessed on 8 April 2022).
- Blankenberg, D.; Von Kuster, G.; Bouvier, D.; Baker, D.; Afgan, E.; Stoler, N.; Taylor, J.; Nekrutenko, A.; Galaxy the Galaxy Team. Dissemination of scientific software with Galaxy ToolShed. Genome Biol. 2014, 15, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 27 March 2022).
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Madan, A. CAP3: A DNA Sequence Assembly Program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Blast-Tools. Available online: https://github.com/schmidda/blast-tools (accessed on 27 March 2022).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2015, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Galaxy-Tools. Available online: https://github.com/bgruening/galaxytools (accessed on 27 March 2022).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2013, 42, D7–D17. [Google Scholar]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource–improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef]
- Ganten, D.; Ruckpaul, K. Batch Entrez. In Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine; Springer: Berlin, Germany, 2006; p. 131. [Google Scholar]
- Gutiérrez, P.; Rivillas, A.; Tejada, D.; Giraldo, S.; Restrepo, A.; Ospina, M.; Marín, M. PVDP: A portable open source pipeline for detection of plant viruses in RNAseq data. A case study on potato viruses in Antioquia (Colombia). Physiol. Mol. Plant Pathol. 2021, 113, 101604. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Wright, C.; Rajpurohit, A.; Burke, E.E.; Williams, C.; Collado-Torres, L.; Kimos, M.; Brandon, N.J.; Cross, A.J.; Jaffe, A.E.; Weinberger, D.R.; et al. Comprehensive assessment of multiple biases in small RNA sequencing reveals significant differences in the performance of widely used methods. BMC Genom. 2019, 20, 513. [Google Scholar] [CrossRef] [Green Version]
- International Committee of Taxonomy of Viruses (ICTV). VMR File Repository—VMR. Available online: https://talk.ictvonline.org/taxonomy/vmr/m/vmr-file-repository/ (accessed on 30 June 2022).
- Leblanc, Z.; Gauthier, M.-E.; Lelwala, R.; Elliott, C.; McMaster, C.; Eichner, R.; Davis, K.; Liefting, L.; Thompson, J.; Dinsdale, A.; et al. Complete genome sequence of a novel potyvirus infecting Miscanthus sinensis (silver grass). Arch. Virol. 2022, 167, 1–5. [Google Scholar] [CrossRef]
- Césari, S.; Kanzaki, H.; Fujiwara, T.; Bernoux, M.; Chalvon, V.; Kawano, Y.; Shimamoto, K.; Dodds, P.; Terauchi, R.; Kroj, T. The NB-LRR proteins RGA 4 and RGA 5 interact functionally and physically to confer disease resistance. EMBO J. 2014, 33, 1941–1959. [Google Scholar] [CrossRef]
- Wei, L.; Gu, L.; Song, X.; Cui, X.; Lu, Z.; Zhou, M.; Wang, L.; Hu, F.; Zhai, J.; Meyers, B.C.; et al. Dicer-like 3 produces transposable element-associated 24-nt siRNAs that control agricultural traits in rice. Proc. Natl. Acad. Sci. USA 2014, 111, 3877–3882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Serio, F.; Flores, R.; Verhoeven, J.T.J.; Li, S.F.; Pallás, V.; Randles, J.W.; Owens, R.A. Current status of viroid taxonomy. Arch. Virol. 2014, 159, 3467–3478. [Google Scholar] [CrossRef] [PubMed]
- Poojari, S.; Alabi, O.J.; Fofanov, V.Y.; Naidu, R.A. Correction: A Leafhopper-Transmissible DNA Virus with Novel Evolutionary Lineage in the Family Geminiviridae Implicated in Grapevine Redleaf Disease by Next-Generation Sequencing. PLoS ONE 2016, 11, e0147510. [Google Scholar] [CrossRef]
- International Committee of Taxonomy of Viruses (ICTV). Proposal 2020.031P—Abolish the Genus Mandarivirus, Create a New Subgenus (Mandarivirus) in the Genus Potexvirus and One New Subgenus (Acarillexivirus) in the Genus Allexivirus (Tymovorales: Alphaflexiviridae). Available online: https://talk.ictvonline.org (accessed on 30 May 2022).
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef] [PubMed]
- Scheets, W.M.K.; Somera, M. (2020, Abolish the Family Luteoviridae (Tolivirales) and Move Its Genera to the Families Tombusviridae (Tolivirales) and Solemoviridae (Sobelivirales). Available online: https://ictv.global/ictv/proposals/2020.026P.R.Abolish_Luteoviridae.zip (accessed on 30 May 2022).
- Adams, M.J.; Antoniw, J.F. DPVweb: A comprehensive database of plant and fungal virus genes and genomes. Nucleic Acids Res. 2006, 34, D382–D385. [Google Scholar] [CrossRef] [Green Version]
- Bagheri, H.; Severin, A.J.; Rajan, H. Detecting and correcting misclassified sequences in the large-scale public databases. Bioinformatics 2020, 36, 4699–4705. [Google Scholar] [CrossRef]
- Honjo, M.N.; Emura, N.; Kawagoe, T.; Sugisaka, J.; Kamitani, M.; Nagano, A.J.; Kudoh, H. Seasonality of interactions between a plant virus and its host during persistent infection in a natural environment. ISME J. 2020, 14, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Alcaide, C.; Sardanyés, J.; Elena, S.F.; Gómez, P. Increasing temperature alters the within-host competition of viral strains and influences virus genetic variability. Virus Evol. 2021, 7, veab017. [Google Scholar] [CrossRef]
- Hipper, C.; Brault, V.; Ziegler-Graff, V.; Revers, F. Viral and Cellular Factors Involved in Phloem Transport of Plant Viruses. Front. Plant Sci. 2013, 4, 154. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, G.; Zwart, M.P.; Elena, S.F. Onset of virus systemic infection in plants is determined by speed of cell-to-cell movement and number of primary infection foci. J. R. Soc. Interface 2014, 11, 20140555. [Google Scholar] [CrossRef]
- Illumina, I. Effects of Index Misassignment on Multiplexing and Downstream Analysis. Available online: www.illumina.com (accessed on 30 May 2022).
- Costello, M.; Fleharty, M.; Abreu, J.; Farjoun, Y.; Ferriera, S.; Holmes, L.; Gabriel, S. Characterization and remediation of sample index swaps by non-redundant dual indexing on massively parallel sequencing platforms. BMC Genom. 2018, 19, 332. [Google Scholar] [CrossRef] [PubMed]
- MacConaill, L.E.; Burns, R.T.; Nag, A.; Coleman, H.A.; Slevin, M.K.; Giorda, K.; Thorner, A.R. Unique, dual-indexed sequencing adapters with UMIs effectively eliminate index cross-talk and significantly improve sensitivity of massively parallel sequencing. BMC Genom. 2018, 19, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouché, N.; Lauressergues, D.; Gasciolli, V.; Vaucheret, H. An antagonistic function for Arabidopsis DCL2 in development and a new function for DCL4 in generating viral siRNAs. EMBO J. 2006, 25, 3347–3356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deleris, A.; Gallego-Bartolome, J.; Bao, J.; Kasschau, K.D.; Carrington, J.C.; Voinnet, O. Hierarchical Action and Inhibition of Plant Dicer-Like Proteins in Antiviral Defense. Science 2006, 313, 68–71. [Google Scholar] [CrossRef]
- Xie, Z.; Johansen, L.K.; Gustafson, A.M.; Kasschau, K.D.; Lellis, A.D.; Zilberman, D.; Weigel, D. Genetic and Functional Diversification of Small RNA Pathways in Plants. PLOS Biol. 2004, 2, e104. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Xue, Y.; Zhang, L.; Zhong, Z.; Feng, S.; Wang, C.; Xiao, L.; Yang, Z.; Harris, C.J.; Wu, Z.; et al. Mechanism of siRNA production by a plant Dicer-RNA complex in dicing-competent conformation. Science 2021, 374, 1152–1157. [Google Scholar] [CrossRef]
- Deng, Z.; Ma, L.; Zhang, P.; Zhu, H. Small RNAs Participate in Plant–Virus Interaction and Their Application in Plant Viral Defense. Int. J. Mol. Sci. 2022, 23, 696. [Google Scholar] [CrossRef]
- Dunoyer, P. La bataille du silence. Med. Sci. 2009, 25, 505–512. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A Greedy Algorithm for Aligning DNA Sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Parker, V.L.; Cushen, B.F.; Gavriil, E.; Marshall, B.; Waite, S.; Pacey, A.; Heath, P.R. Comparison and optimisation of microRNA extraction from the plasma of healthy pregnant women. Mol. Med. Rep. 2021, 23, 258. [Google Scholar] [CrossRef]
- Kesanakurti, P.; Belton, M.; Saeed, H.; Rast, H.; Boyes, I.; Rott, M. Screening for plant viruses by next generation sequencing using a modified double strand RNA extraction protocol with an internal amplification control. J. Virol. Methods 2016, 236, 35–40. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Commodity | Species | Known PEQ Detections 1 | Detections from VirReport 2 | Detections from GA-VirReport 3 |
|---|---|---|---|---|---|
| MT001 | Citrus | Citrus Troyer × Frost-Lisbon | CEVd | CEVd | CEVd |
| MT002 | Stonefruit | Prunus persica (Nectarine) | PNRSV | PNRSV | PNRSV |
| MT003 | Citrus | Citrus aurantiifolia (Christm.) Swingle | CTV, CVEV, CDVd, HSVd | CTV, CVEV, CDVd, HSVd | CTV, CVEV, CDVd, HSVd |
| MT005 | Raspberry | Rubus idaeus | RBDV | RBDV, RYNV | RBDV, RYNV |
| MT008 | Citrus | Citrus sinensis | CDVd, HSVd | CDVd, HSVd, CiVA | CDVd, HSVd, CiVA |
| MT010 | Ornamental grass | Miscanthus sinensis ‘Morning light’ | Novel Potyvirus (MsiMV) | Novel Potyvirus (MsiMV) | Novel Potyvirus (MsiMV) |
| MT012 | Iris | Iris sp. ‘Crimson colossus’ | ISMV, TRSV | ISMV, TRSV, CRLV | ISMV, TRSV, CRLV |
| MT016 | Sweet Potato | Ipomoea batatas ‘GRF0334’ | SPFMV | SPFMV, SPSMV-1, SPBV | SPFMV, SPSMV-1, SPBV |
| Sample ID | Commodity | Species | 1 GA-VirReport Detection | 2 Subject Accession | 3 Average Percent Identity | 4 Percentage Contig Coverage | 5 Percentage Bases 10× Read Depth |
|---|---|---|---|---|---|---|---|
| MT338 | Stone fruit | Prunus salicina | PrLV | MF510412 | 100 | 41.26 | 85.54% |
| MT341 | Stone fruit | Prunus salicina | PrLV | MF510412 | 100 | 25.95 | 66.43% |
| MT350 | Stone fruit | Prunus avium | CVA | LC422952 | 99.41 | 32.41 | 79.78% |
| PDV | MT013233 | 98.8 | 64.01 | 96.00% | |||
| MT357 | Stone fruit | Prunus salicina × armeniaca | NeVM | KT273412 | 99.48 | 47.71 | 95.01% |
| MT362 | Stone fruit | Prunus persica var. nucipersica | NSPaV | KT273410 | 99.15 | 76.86 | 98.24% |
| MT363 | Stone fruit | Prunus persica var. nucipersica | NSPaV | KT273410 | 99.06 | 76.32 | 97.44% |
| MT365 | Stone fruit | Prunus persica var. nucipersica | NeVM | KT273412 | 99.16 | 60.24 | 96.51% |
| NSPaV | KT273410 | 99.29 | 77.84 | 98.16% | |||
| MT368 | Stone fruit | Prunus persica var. nucipersica | NeVM | KT273412 | 98.61 | 50.19 | 96.83% |
| MT370 | Stone fruit | Prunus persica | NSPaV | KT273410 | 99.34 | 68.48 | 96.31% |
| MT378 | Stone fruit | Prunus salicinia × avium | NeVM | KT273412 | 99.03 | 56.28 | 97.82% |
| MT383 | Stone fruit | Prunus avium | CVA | KX370827 | 98.19 | 38.27 | 95.67% |
| PDV | MT013233 | 98.81 | 74.61 | 97.96% | |||
| MT385 | Stone fruit | Prunus avium | CVA | LC523004 | 92.44 | 14.1 | 98.07% |
| PDV | MT013233 | 98.75 | 72.99 | 79.58% | |||
| MT410 | Apple | Malus domestica | CCGaV | MK940543 | 99.79 | 33.56 | 76.70% |
| ASPV | KF321967 | 96.87 | 24.55 | 59.02% | |||
| ACLSV | KR606325 | 98.04 | 47.2 | 97.31% | |||
| MT411 | Grapevine | Vitis vinifera | HSVd | MF979532 | 100 | 51.34 | 100.00% |
| GRSPaV | MG938345 | 98.19 | 43.42 | 90.52% | |||
| GYSVd | DQ377130 | 100 | 34.9 | 96.12% | |||
| AGVd | GU327604 | 100 | 14.68 | 79.82% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lelwala, R.V.; LeBlanc, Z.; Gauthier, M.-E.A.; Elliott, C.E.; Constable, F.E.; Murphy, G.; Tyle, C.; Dinsdale, A.; Whattam, M.; Pattemore, J.; et al. Implementation of GA-VirReport, a Web-Based Bioinformatics Toolkit for Post-Entry Quarantine Screening of Virus and Viroids in Plants. Viruses 2022, 14, 1480. https://doi.org/10.3390/v14071480
Lelwala RV, LeBlanc Z, Gauthier M-EA, Elliott CE, Constable FE, Murphy G, Tyle C, Dinsdale A, Whattam M, Pattemore J, et al. Implementation of GA-VirReport, a Web-Based Bioinformatics Toolkit for Post-Entry Quarantine Screening of Virus and Viroids in Plants. Viruses. 2022; 14(7):1480. https://doi.org/10.3390/v14071480
Chicago/Turabian StyleLelwala, Ruvini V., Zacharie LeBlanc, Marie-Emilie A. Gauthier, Candace E. Elliott, Fiona E. Constable, Greg Murphy, Callum Tyle, Adrian Dinsdale, Mark Whattam, Julie Pattemore, and et al. 2022. "Implementation of GA-VirReport, a Web-Based Bioinformatics Toolkit for Post-Entry Quarantine Screening of Virus and Viroids in Plants" Viruses 14, no. 7: 1480. https://doi.org/10.3390/v14071480
APA StyleLelwala, R. V., LeBlanc, Z., Gauthier, M.-E. A., Elliott, C. E., Constable, F. E., Murphy, G., Tyle, C., Dinsdale, A., Whattam, M., Pattemore, J., & Barrero, R. A. (2022). Implementation of GA-VirReport, a Web-Based Bioinformatics Toolkit for Post-Entry Quarantine Screening of Virus and Viroids in Plants. Viruses, 14(7), 1480. https://doi.org/10.3390/v14071480