
A Deeper Insight into Evolutionary Patterns and Phylogenetic History of ORF Virus through the Whole Genome Sequencing of the First Italian Strains

 ,
,  ,
,  ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Virus Isolation
2.3. Viral DNA Extraction, Sequencing and Genome Assembly
2.4. Phylogeny, Molecular Dating and Evolutionary Rate
3. Results
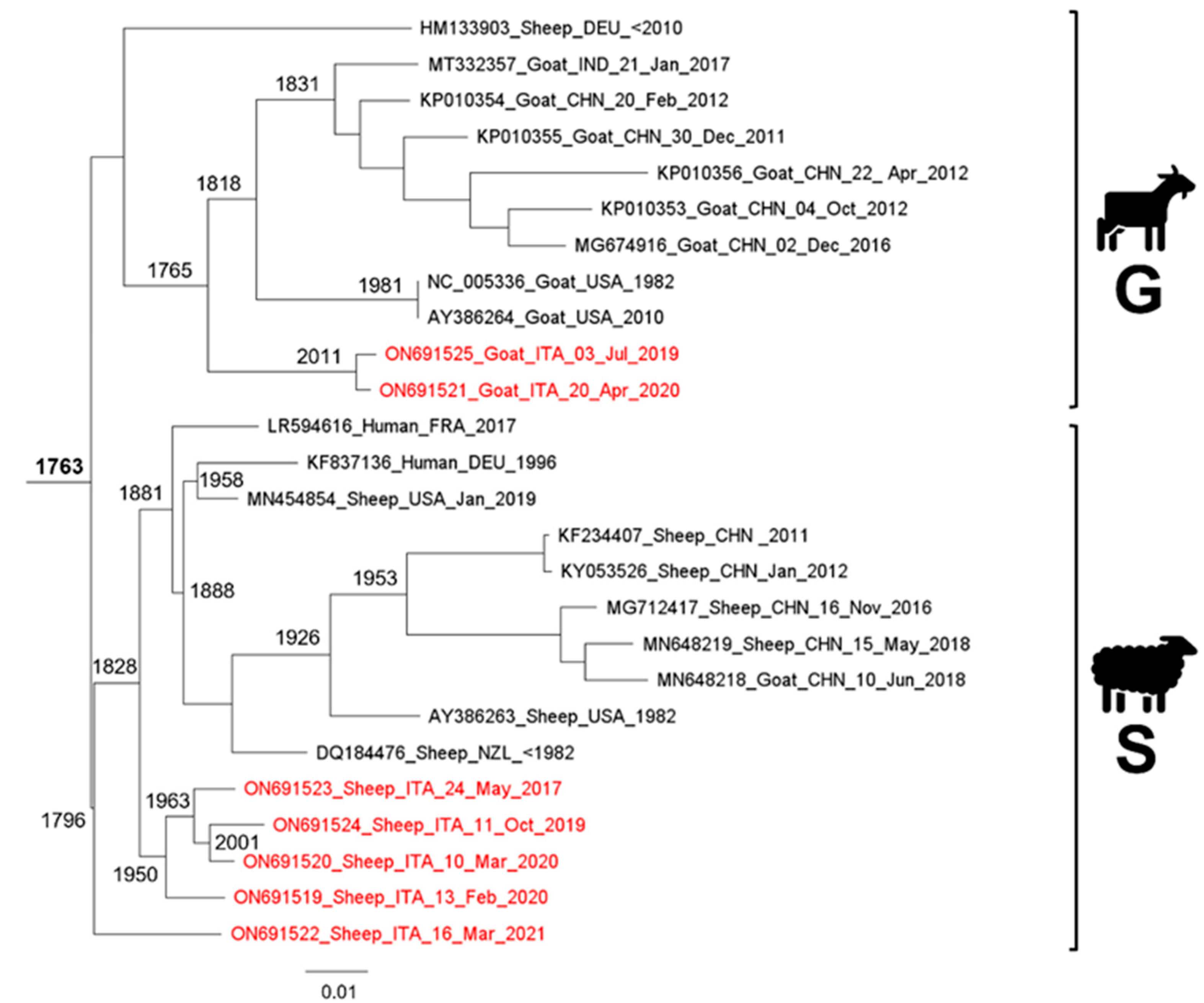
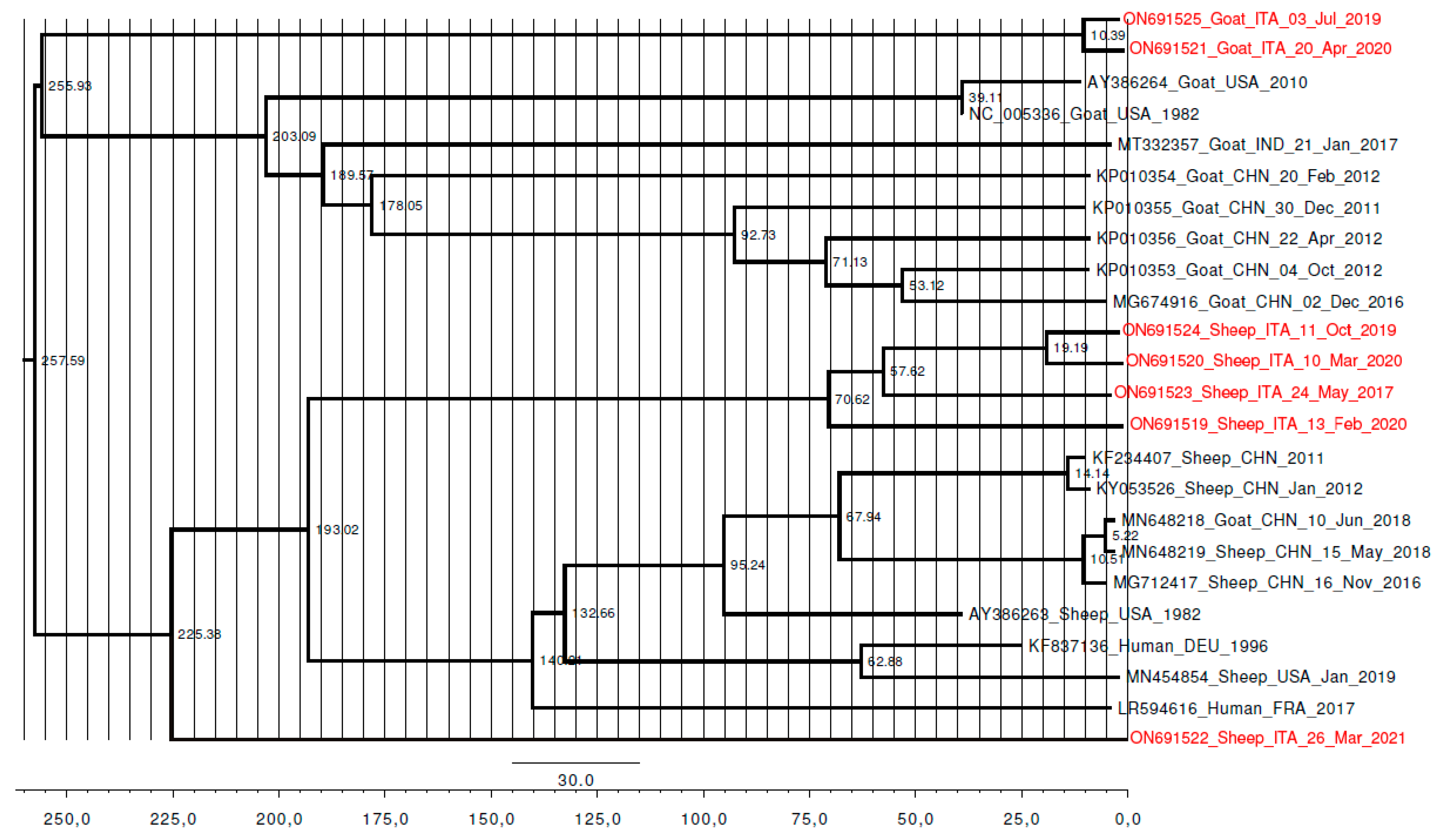
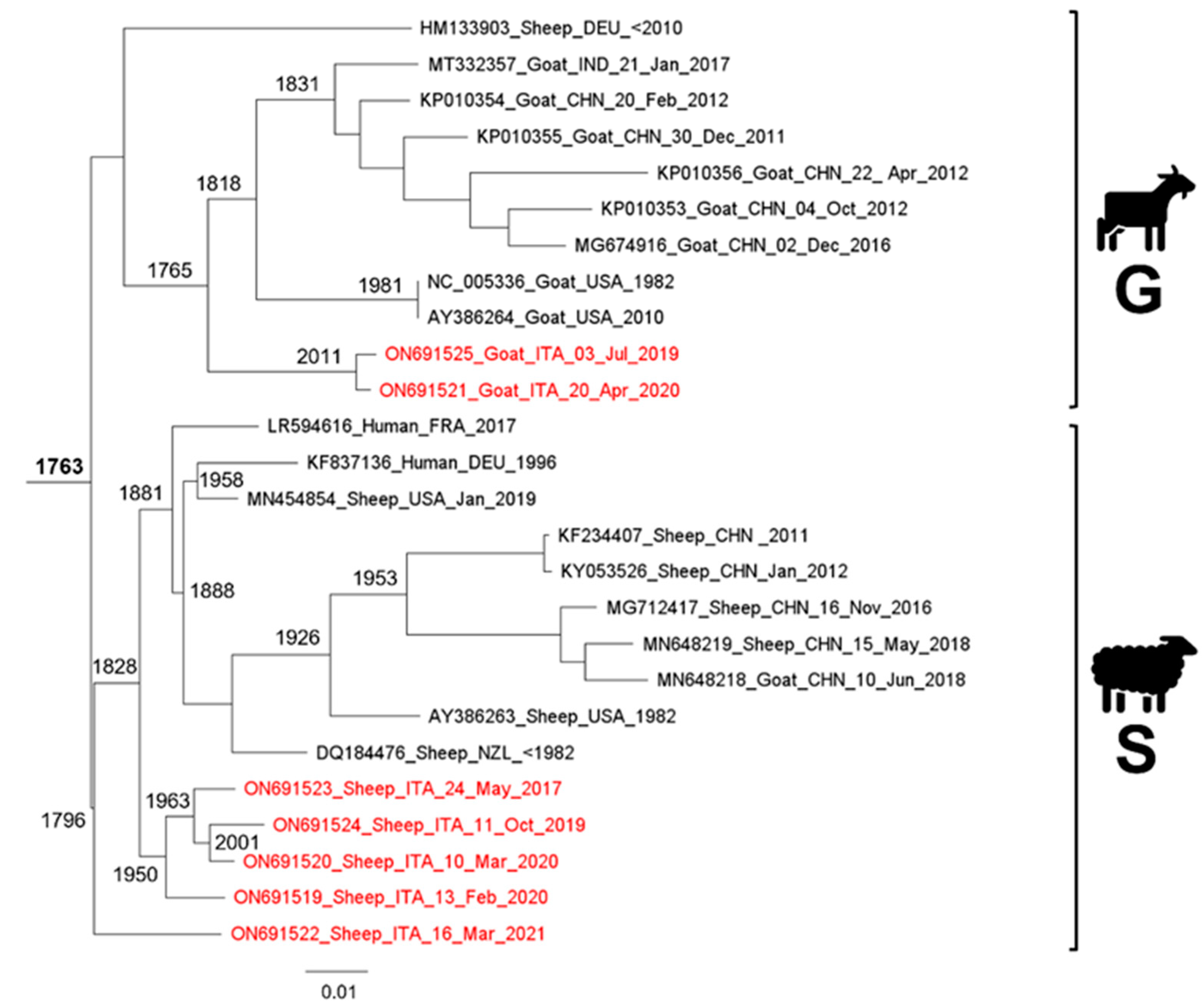
Phylogeny, Molecular Dating, and Evolutionary Rate
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| S6 | S10 | S15 | S19 | S21 | S27 | S30 | ||
|---|---|---|---|---|---|---|---|---|
| Culture isolate | Sheep crusted lesion | Lamb crusted lesion | Kid proliferative lesion | Lamb crusted lesion | Sheep crusted lesion | Sheep crusted lesion | Kid crusted lesion | |
| Genome length | 128,110 nt | 128,257 nt | 129,776 nt | 131,660 nt | 129,506 nt | 129,858 nt | 130,411 nt | |
| n° High quality reads mapped | 1,243,066 | 7959 | 4681 | 277,763 | 6570 | 25,512 | 300,184 | |
| % Mapped genome | 91.5 | 91.6 | 92.7 | 94.1 | 92.5 | 92.8 | 93.2 | |
| Average per base coverage | 1453.3× | 9.3× | 16.9× | 315× | 7.6× | 31.6× | 343.3× | |
| Base composition | A: 17.6% | A: 17.8% | A: 17.8% | A: 17.8% | A: 17.8% | A: 17.8% | A: 17.9% | |
| C: 32.5% | C: 32.3% | C: 32.2% | C: 32.2% | C: 32.3% | C: 32.3% | C: 32.3% | ||
| G: 32.6% | G: 32.2% | G: 32.2% | G: 32.2% | G: 32.2% | G: 32.3% | G: 32.3% | ||
| T: 17.3% | T: 17.7% | T: 17.7% | T: 7.7% | T: 17.6% | T: 17.6% | T: 17.5% | ||
| Contigs information * | Max length | 69,576 nt | 5175 nt | 8125 nt | 69,407 nt | 8126 nt | 9279 nt | 488,393 nt |
| Min length | 104 nt | 112 nt | 92 nt | 92 nt | 92 nt | 92 nt | 92 nt | |
| Total number | 184 contigs | 2245 contigs | 778 contigs | 1041 contigs | 746 contigs | 4474 contigs | 1022 contigs | |
| % Identity with the reference genome NC_005336 ** | 97% | 98% | 98% | 97% | 97% | 97% | 98% | |
| Sample ID | GenBank | Biosample | Sequence Read Archive (SRA) | BioProject |
|---|---|---|---|---|
| S21 | ON691523 | SAMN28405497 | SRR19732912 | PRJNA838097 |
| S30 | ON691525 | SAMN28405499 | SRR19732910 | |
| S27 | ON691524 | SAMN28405498 | SRR19732911 | |
| S6 | ON691519 | SAMN28405493 | SRR19732916 | |
| S10 | ON691520 | SAMN28405494 | SRR19732915 | |
| S15 | ON691521 | SAMN28405495 | SRR19732914 | |
| S19 | ON691522 | SAMN28405496 | SRR19732913 |
References
- Bergqvist, C.; Kurban, M.; Abbas, O. Orf virus infection. Rev. Med. Virol. 2017, 27, e1932. [Google Scholar] [CrossRef]
- Schmidt, D. Die dermatitis pustulosa des Scafes. (Dermatitis pustulosa in sheep). In Handbuch der Virusinfektionen bei Tieren; Rehner, H., Ed.; Gustav Fischer Verlag: Jena, Germany, 1967; pp. 713–761. [Google Scholar]
- Hansen. Orf in London. Norge Tidskr. Vet. 1879, 9, 298. [Google Scholar]
- Berry, A.H. Contagious pustular dermatitis of sheep. J. Comp. Pathol. Ther. 1901, 14, 307–312. [Google Scholar] [CrossRef]
- Howarth, J.A. Infectious pustolar dermatitis of sheep and goats. J. Am. Vet. Med. Assoc. 1929, 29, 741–761. [Google Scholar]
- Boughton, I.B.; Hardy, W.T. Infectious entero-toxemia (milk colic) of lambs and kids. TAES—Bull. 1941, 598, 20. [Google Scholar]
- Hosamani, M.; Scagliarini, A.; Bhanuprakash, V.; McInnes, C.J.; Singh, R.K. Orf: An update on current research and future perspectives. Expert Rev. Anti. Infect. Ther. 2009, 7, 879–893. [Google Scholar] [CrossRef]
- Vaccari, F. Meccanismi Evolutivi dei Parapoxvirus: Caratterizzazione Genomica di Pseudocowpoxvirus e Messa a Punto di Sistemi per lo Studio delle Ricombinazioni. Ph.D. Thesis, University of Bologna, Bologna, Italy, 2009. [Google Scholar]
- Kumar, R.; Trivedi, R.N.; Bhatt, P.; Khan, S.H.; Khurana, S.K.; Tiwari, R.; Karthik, K.; Malik, Y.S.; Dhama, K.; Chandra, R. Contagious pustular dermatitis (orf disease)—Epidemiology, diagnosis, control and public health concerns. Adv. Anim. Vet. Sci. 2015, 3, 649–676. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhu, X.; Zheng, Y.; Wang, S.; Liu, Z.; Dou, Y.; Li, H.; Cai, X.; Luo, X. Phylogenetic analysis of two Chinese orf virus isolates based on sequences of B2L and VIR genes. Arch. Virol. 2013, 158, 1477–1485. [Google Scholar] [CrossRef]
- Gumbrell, R.C.; McGregor, D.A. Outbreak of severe fatal orf in lambs. Vet. Rec. 1997, 141, 150–151. [Google Scholar] [CrossRef]
- Mazur, C.; Machado, R.D. The isolation and identification of the contagious ecthyma virus of caprines in cell cultures. Rev. Microbiol. Sao Paulo 1990, 21, 127–130. [Google Scholar]
- Mazur, C.; Machado, R.D. Detection of contagious pustular dermatitis virus of goats in a severe outbreak. Vet. Rec. 1998, 125, 419–420. [Google Scholar] [CrossRef]
- Nandi, S.; De, U.K.; Chowdhury, S. Current status of contagious ecthyma or orf disease in goat and sheep—A global perspective. Small Rumin. Res. 2011, 96, 73–82. [Google Scholar] [CrossRef]
- McInnes, C.J. Orf. Vet. Dermatol. 2014, 25, 341–342. [Google Scholar] [CrossRef]
- Spyrou, V.; Valiakos, G. Orf virus infection in sheep or goats. Vet. Microbiol. 2015, 181, 178–182. [Google Scholar] [CrossRef]
- Li, W.; Hao, W.; Peng, Y.; Duan, C.; Tong, C.; Song, D.; Gao, F.; Li, M.; Rock, D.L.; Luo, S. Comparative genomic sequence analysis of Chinese orf virus strain NA1/11 with other parapoxviruses. Arch. Virol. 2015, 160, 253–266. [Google Scholar] [CrossRef]
- Fleming, S.B.; Wise, L.M.; Mercer, A.A. Molecular Genetic Analysis of Orf Virus: A Poxvirus That Has Adapted to Skin. Viruses 2015, 7, 1505–1539. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Y.; Liu, F.; Shuhong, L. Orf virus: A promising new therapeutic agent. Rev. Med. Virol. 2018, 29, e2013. [Google Scholar] [CrossRef] [Green Version]
- Chi, X.; Zeng, X.; Li, W.; Hao, W.; Li, M.; Huang, X.; Rock, D.L.; Luo, S.; Wang, S. Genome analysis of orf virus isolates from goats in the Fujian Province of southern China. Front. Microbiol. 2015, 6, 1135. [Google Scholar] [CrossRef]
- Andreani, J.; Fongue, J.; Khalil, J.Y.B.; David, L.; Mougari, S.; Le Bideau, M.; Abrahão, J.; Berbis, P.; La Scola, B. Human infection with orf virus and description of its whole genome, France. Emerg. Infect. Dis. 2017, 25, 2197. [Google Scholar] [CrossRef]
- Coradduzza, E.; Sanna, D.; Rocchigiani, A.M.; Pintus, D.; Scarpa, F.; Scivoli, R.; Bechere, R.; Dettori, M.A.; Montesu, M.A.; Marras, V.; et al. Molecular insights into the genetic variability of ORF virus in a Mediterranean region (Sardinia, Italy). Life 2021, 11, 416. [Google Scholar] [CrossRef]
- Fiori, M.S.; Sanna, D.; Scarpa, F.; Floris, M.; Di Nardo, A.; Ferretti, L.; Loi, F.; Cappai, S.; Sechi, A.M.; Angioi, P.P.; et al. Deeper Insight into Evolutionary Patterns and Phylogenetic History of ASFV Epidemics in Sardinia (Italy) through Extensive Genomic Sequencing. Viruses 2021, 13, 1994. [Google Scholar] [CrossRef]
- Hannon, G.J. FASTX-Toolkit 2010. Available online: http://hannonlab.cshl.edu/fastx_toolkit (accessed on 26 May 2022).
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Scarpa, F.; Sanna, D.; Azzena, I.; Cossu, P.; Mugetti, D.; Cerutti, F.; Prearo, M.; Peletto, S.; Casu, M. Draft genome sequence of Rhodococcus qingshengii strain PN_19, isolated from a moribund individual of Pinna nobilis in Sardinia, Italy. Microbiol. Resour. Announc. 2021, 10, e00356-21. [Google Scholar] [CrossRef]
- Piredda, I.; Scarpa, F.; Sanna, D.; Casu, M.; Ponti, M.N.; Chisu, V. Draft genome sequences of four different strains belonging to Leptospira interrogans serovar Pomona isolated from mammals in the island of Sardinia, Italy. Microbiol. Resour. Announc. 2021, 10, e0069821. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Higgins, D.G. Clustal Omega, Accurate Alignment of Very Large Numbers of Sequences. In Multiple Sequence Alignment Methods; Russell, D., Ed.; Humana Press: Totowa, NJ, USA, 2014; Volume 1079, pp. 105–116. [Google Scholar]
- Nayak, D.; Sahu, B.; Majee, P.; Singh, R.; Sahoo, N. Recombination Drives Emergence of Orf Virus Diversity: Evidence from the First Complete Genome of Indian Orf Virus and Comparative Genomic Analysis. Res. Sq. 2021; in preprint. [Google Scholar] [CrossRef]
- Mercer, A.A.; Ueda, N.; Friederichs, S.M.; Hofmann, K.; Fraser, K.M.; Bateman, T.; Fleming, S.B. Comparative analysis of genome sequences of three isolates of Orf virus reveals unexpected sequence variation. Virus Res. 2006, 116, 146–158. [Google Scholar] [CrossRef]
- Heare, D.L.; Little, S.V.; Weise, D.W.; Harris, J.R.; Hillhouse, A.E.; Konganti, K.; Lawon, S.D. Whole-genome sequence of an orf virus isolate derived from a cell culture infected with contagious ecthyma vaccine. Microbiol. Resour. Announc. 2020, 9, e00752-20. [Google Scholar] [CrossRef]
- Delhon, G.; Tulman, E.R.; Afonso, C.L.; Lu, Z.; De la Concha-Bermejillo, A.; Lehmkuhl, H.D.; Piccone, M.E.; Kutish, G.F.; Rock, D.L. Genomes of the parapoxviruses ORF virus and bovine papular stomatitis virus. J. Virol. 2004, 78, 168–177. [Google Scholar] [CrossRef] [Green Version]
- McGuire, M.J.; Johnston, S.A.; Sykes, K.F. Novel immune-modulator identified by a rapid, functional screen of the parapoxvirus ovis (Orf virus) genome. Proteome Sci. 2012, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Friederichs, S.; Krebs, S.; Blum, H.; Wolf, E.; Lang, H.; Von Buttlar, H.; Buttner, M. Comparative and retrospective molecular analysis of Parapoxvirus (PPV) isolates. Virus Res. 2014, 181, 11–21. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in excel. Population genetic software for teaching and research—An update. Oxf. J. 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Gelman, A.; Rubin, D.B. Inference from iterative simulation using multiple sequences. Stat. Sci. 1992, 7, 457–472. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Cossu, P.; Lai, T.; Casu, M.; Curini-Galletti, M. How to achieve internal fertilization without a vagina: The study case of the genus Archilina Ax, 1959 (Platyhelminthes, Proseriata) from Canary Islands. Mar. Biodiv. 2019, 49, 2057–2073. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, e214. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 645 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Samuel, W.M.; Chalmers, G.A.; Stelfox, J.G.; Loewen, A.; Thomsen, J.J. Contagious ecthyma in bighorn sheep and mountain goat in western Canada. J. Wildl. Dis. 1975, 11, 26–31. [Google Scholar] [CrossRef]
- Teshale, A.; Alemayehu, A. Contagious Ecthyma and its Public Health Significance. Dairy Vet. Sci. J. 2018, 7, 555711. [Google Scholar] [CrossRef] [Green Version]
- Spickler, A.R. Contagious Ecthyma. 2015. Available online: http://www.cfsph.iastate.edu/DiseaseInfo/factsheets.php (accessed on 25 May 2022).
- Scarpa, F.; Casu, M.; Sanna, D. Evolutionary and Conservation Genetics. Life 2021, 11, 1160. [Google Scholar] [CrossRef]
- Sackman, A.M.; McGee, L.W.; Morrison, A.J.; Pierce, J.; Anisman, J.; Hamilton, H.; Sanderbeck, S.; Newman, C.; Rokyta, D.R. Mutation-Driven Parallel Evolution during Viral Adaptation. Mol. Biol. Evol. 2017, 34, 3243–3253. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, S.R.; Patel, M.R. Chronic Infections. In Green’s Operative Hand Surgery, 8th ed.; Wolfe, S.W., Pederson, W.C., Kozin, S.H., Cohen, M.S., Eds.; Elsevier Health Sciences: Philadelphia, PA, USA, 2021; pp. 62–127. [Google Scholar]
- Aynaud, M. La Stomatite pustuleuse contagieuse des virus. (Chancre du mouton). Ann. Inst. Pasteur. 1923, 37, 498. [Google Scholar]
- Theiler, A. Ecthyma Contagiosum of Sheep and Goats; 13th and 14th reports of the Director of Veter; Government Printer and Stationery Office: Pretoria, South Africa, 1928.
- Hatziolos, B.S.C. L’Ecthyma Contagieux du Mouton. Doctoral Dissertation, Jouve Group, Paris, France, 1929. [Google Scholar]

| Sample ID | GB # | Country | Collection Site | Co-Ordinates | Collection Date | Host |
|---|---|---|---|---|---|---|
| S21 | ON691523 | Italy | Sardinia-Sassari | LAT: 40.77916 LON: 8.41332 | 24 May 2017 | Sheep |
| S30 | ON691525 | Italy | Sardinia-Sassari | LAT: 40.60107 LON: 8.34540 | 3 July 2019 | Goat |
| S27 | ON691524 | Italy | Sardinia-Sassari | LAT: 40.76113 LON: 9.0016 | 11 October 2019 | Sheep |
| S6 | ON691519 | Italy | Sardinia-Sassari | LAT: 40.60107 LON: 9.0016 | 13 February 2020 | Sheep |
| S10 | ON691520 | Italy | Sardinia-Sassari | LAT: 40.58005 LON: 9.09239 | 10 March 2020 | Sheep |
| S15 | ON691521 | Italy | Sardinia-Sassari | LAT: 40.64278 LON: 8.89063 | 20 April 2020 | Goat |
| S19 | ON691522 | Italy | Sardinia-Cagliari | LAT: 39.28786 LON: 9.25448 | 26 March 2021 | Sheep |
| Sample ID | GB # | Country | Collection Date | Host | Reference |
|---|---|---|---|---|---|
| NA1/11 | KF234407 | China | 26 October 2011 | Sheep | Li et al., 2013 [10] |
| OVHN3/12 | KY053526 | China | January 2012 | Sheep | Not available |
| SY17 | MG712417 | China | 16 November 2016 | Sheep | Not available |
| CL18 | MN648219 | China | 15 May 2018 | Sheep | Not available |
| NP | KP010355 | China | 30 December 2011 | Goat | Chi et al., 2015 [20] |
| GO | KP010354 | China | 20 February 2012 | Goat | Chi et al., 2015 [20] |
| SJ1 | KP010356 | China | 22 April 2012 | Goat | Chi et al., 2015 [20] |
| YX | KP010353 | China | 04 October 2012 | Goat | Chi et al., 2015 [20] |
| NA17 | MG674916 | China | 02 December 2016 | Goat | Not available |
| GZ18 | MN648218 | China | 10 June 2018 | Goat | Not available |
| MP | MT332357 | India | 21 January 2017 | Goat | Nayak et al., 2020 [32] |
| NZ2 | DQ184476 | New Zealand | Unknown ≤ 1982 | Sheep | Mercer et al., 2006 [33] |
| TVL | MN454854 | USA | January 2019 | Sheep | Heare et al., 2019 [34] |
| OV-IA82 | AY386263 | USA | 1982 | Sheep | Delhon et al., 2004 [35] |
| OV-SA00 | NC005336 | USA | 1982 | Goat | Delhon et al., 2004 [35] |
| OV-SA00 | AY386264 | USA | 2010 | Goat | Delhon et al., 2004 [35] |
| IHUMI-1 | LR594616 | France | 2017 | Human | Andreani et al., 2019 [21] |
| D1701 | HM133903 | Germany | Unknown ≤ 2010 | Sheep | McGuire et al., 2010 [36] |
| B029 | KF837136 | Germany | 1996 | Human | Friederichs et al., 2013 [37] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coradduzza, E.; Sanna, D.; Scarpa, F.; Azzena, I.; Fiori, M.S.; Scivoli, R.; Rocchigiani, A.M.; Bechere, R.; Dettori, M.A.; Pintus, D.; et al. A Deeper Insight into Evolutionary Patterns and Phylogenetic History of ORF Virus through the Whole Genome Sequencing of the First Italian Strains. Viruses 2022, 14, 1473. https://doi.org/10.3390/v14071473
Coradduzza E, Sanna D, Scarpa F, Azzena I, Fiori MS, Scivoli R, Rocchigiani AM, Bechere R, Dettori MA, Pintus D, et al. A Deeper Insight into Evolutionary Patterns and Phylogenetic History of ORF Virus through the Whole Genome Sequencing of the First Italian Strains. Viruses. 2022; 14(7):1473. https://doi.org/10.3390/v14071473
Chicago/Turabian StyleCoradduzza, Elisabetta, Daria Sanna, Fabio Scarpa, Ilenia Azzena, Mariangela S. Fiori, Rosario Scivoli, Angela M. Rocchigiani, Roberto Bechere, Maria A. Dettori, Davide Pintus, and et al. 2022. "A Deeper Insight into Evolutionary Patterns and Phylogenetic History of ORF Virus through the Whole Genome Sequencing of the First Italian Strains" Viruses 14, no. 7: 1473. https://doi.org/10.3390/v14071473
APA StyleCoradduzza, E., Sanna, D., Scarpa, F., Azzena, I., Fiori, M. S., Scivoli, R., Rocchigiani, A. M., Bechere, R., Dettori, M. A., Pintus, D., Evangelista, E., Casu, M., Ligios, C., & Puggioni, G. (2022). A Deeper Insight into Evolutionary Patterns and Phylogenetic History of ORF Virus through the Whole Genome Sequencing of the First Italian Strains. Viruses, 14(7), 1473. https://doi.org/10.3390/v14071473