
A New Circular Single-Stranded DNA Virus Related with Howler Monkey Associated Porprismacovirus 1 Detected in Children with Acute Gastroenteritis

 , ,
, ,  , ,
, , 
 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Information
2.2. Study Population and Sample Collection
2.3. Sample Screening
2.4. Viral-Like Particle Metagenomics
2.5. Bioinformatics Analysis
2.6. Alignments and Annotation
2.7. Genetic Variability
2.8. Phylogenetic Analysis
3. Results
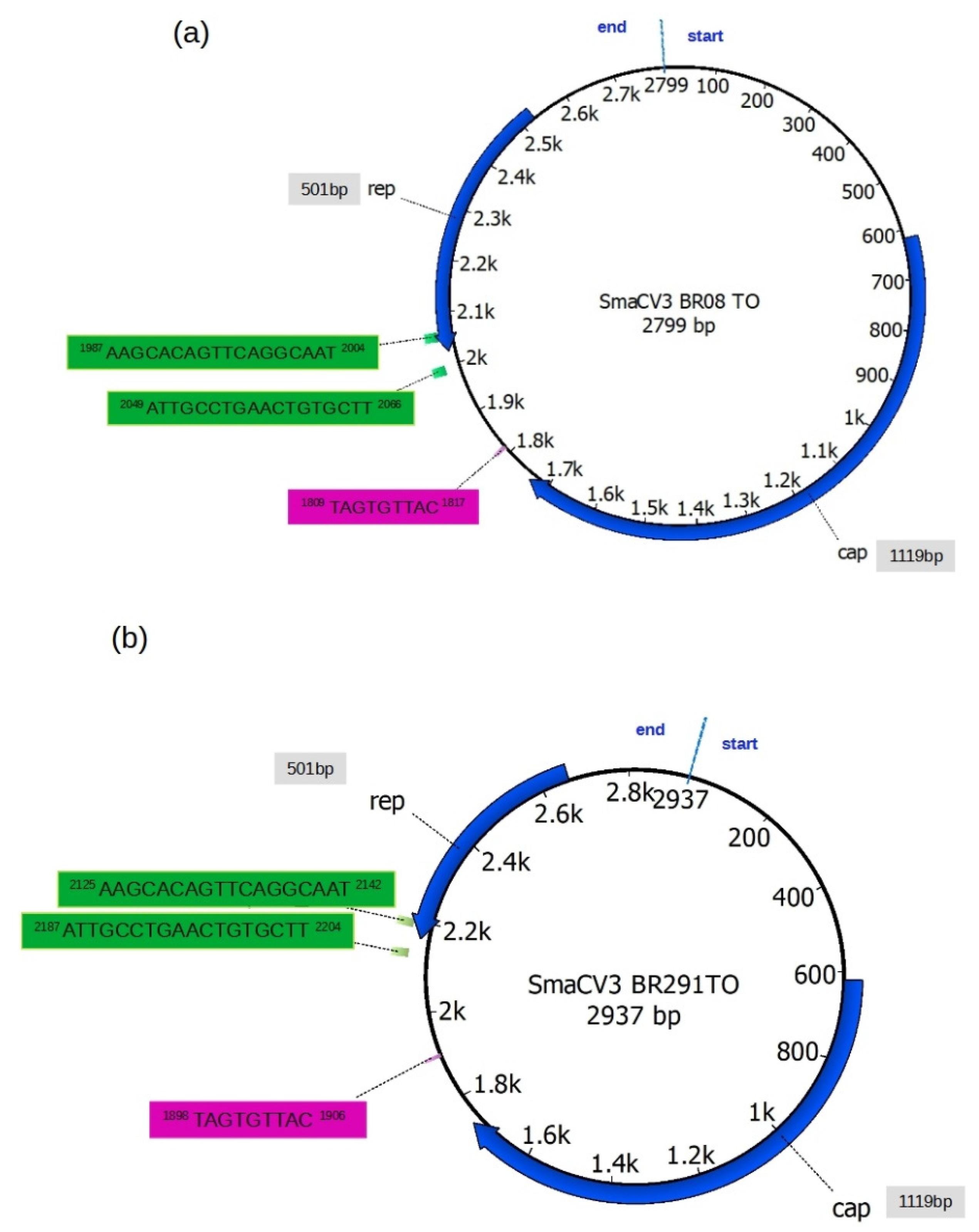
3.1. Blast Search and Sequence Characterization
3.2. Genome Annotation
3.3. Nucleotide Variability in Members of CRESS DNA Viruses
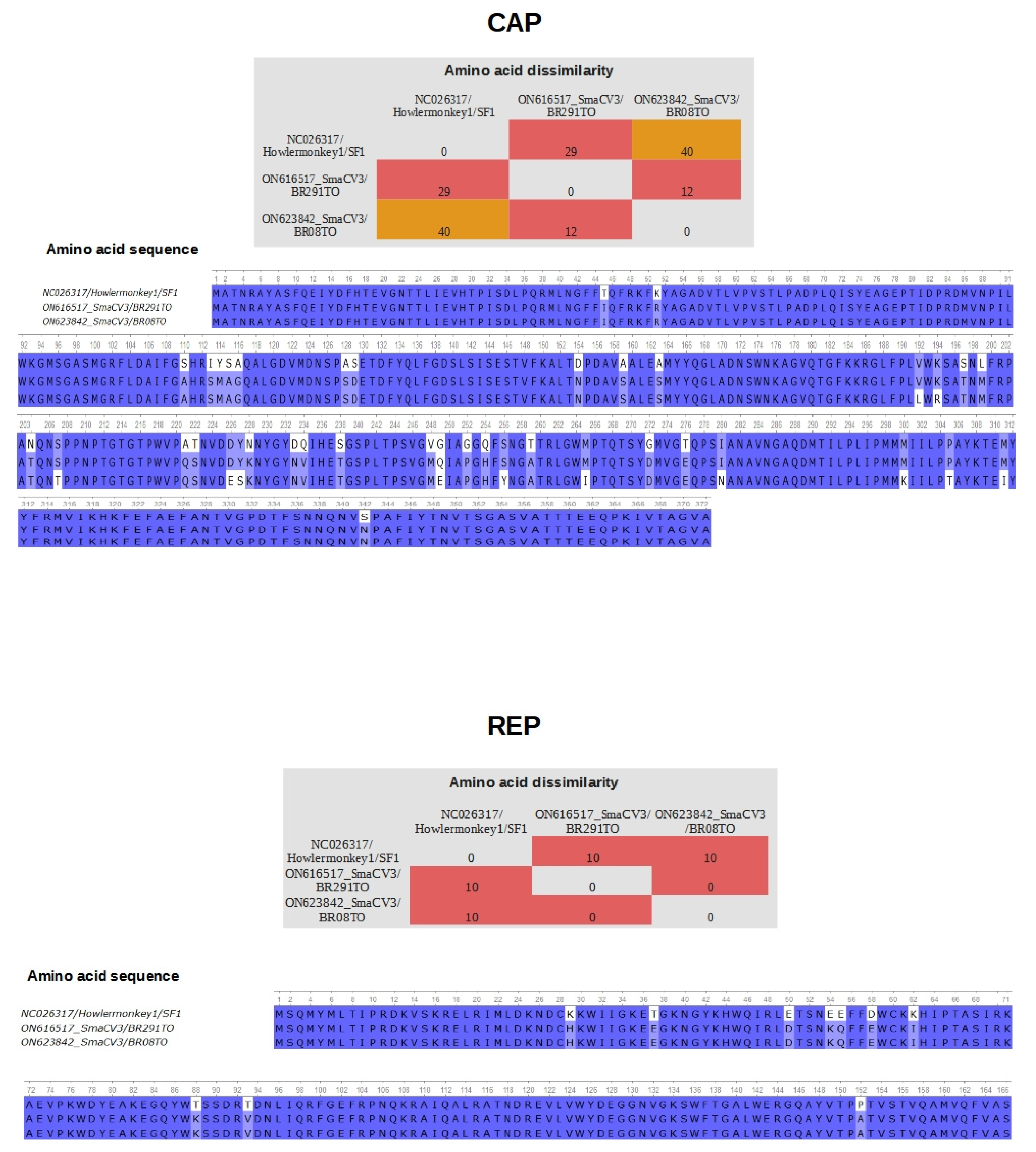
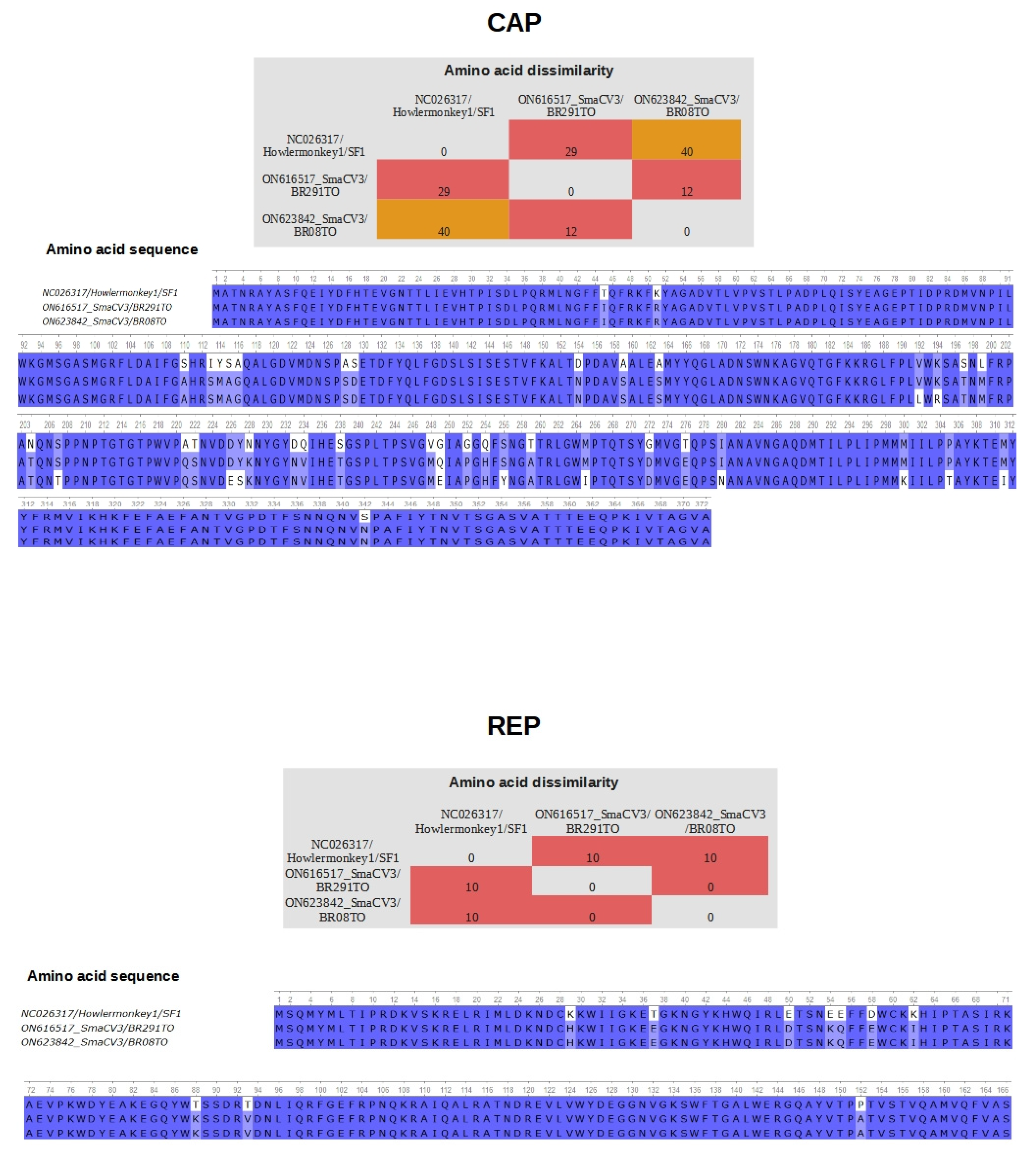
3.4. Differences in Residues in the Proteins CAP and REP of Porprismacovirus
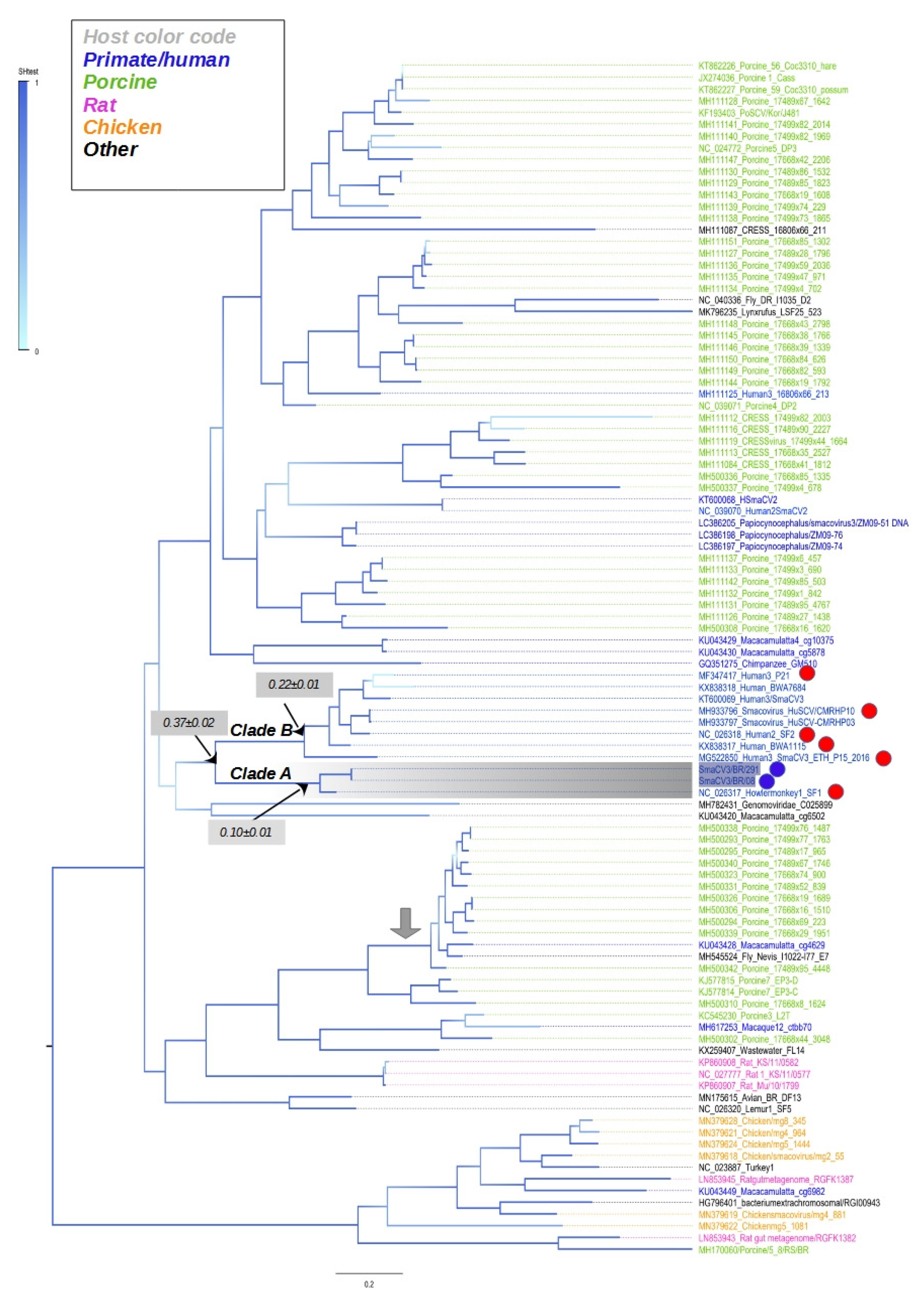
3.5. Phylogenetic Tree of Complete Sequences of CRESS DNA Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosario, K.; Duffy, S.; Breitbart, M. A Field Guide to Eukaryotic Circular Single-Stranded DNA Viruses: Insights Gained from Metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef]
- Zhao, L.; Rosario, K.; Breitbart, M.; Duffy, S. Eukaryotic Circular Rep-Encoding Single-Stranded DNA (CRESS DNA) Viruses: Ubiquitous Viruses with Small Genomes and a Diverse Host Range. Adv. Virus Res. 2019, 103, 71–133. [Google Scholar] [CrossRef]
- Kazlauskas, D.; Varsani, A.; Krupovic, M. Pervasive Chimerism in the Replication-Associated Proteins of Uncultured Single-Stranded DNA Viruses. Viruses 2018, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Wang, H.; Ling, Y.; Yang, S.-X.; Wang, X.-C.; Zhou, R.; Xiao, Y.-Q.; Chen, X.; Yang, J.; Fu, W.-G.; et al. Viral Metagenomics Revealed Diverse CRESS-DNA Virus Genomes in feces of Forest Musk Deer. Virol. J. 2020, 17, 61. [Google Scholar] [CrossRef] [Green Version]
- Chrzastek, K.; Kraberger, S.; Schmidlin, K.; Fontenele, R.S.; Kulkarni, A.; Chappell, L.; Dufour-Zavala, L.; Kapczynski, D.R.; Varsani, A. Diverse Single-Stranded DNA Viruses Identified in Chicken Buccal Swabs. Microorganisms 2021, 9, 2602. [Google Scholar] [CrossRef]
- König, M.-T.; Fux, R.; Link, E.; Sutter, G.; Märtlbauer, E.; Didier, A. Identification and Characterization of Circular Single-Stranded DNA Genomes in Sheep and Goat Milk. Viruses 2021, 13, 2176. [Google Scholar] [CrossRef]
- Shi, Z.; Liu, C.; Yang, H.; Chen, Y.; Liu, H.; Wei, L.; Liu, Z.; Jiang, Y.; He, X.; Wang, J. Fur Seal Feces-Associated Circular DNA Virus Identified in Pigs in Anhui, China. Virol. Sin. 2021, 36, 25–32. [Google Scholar] [CrossRef]
- Kaszab, E.; Lengyel, G.; Marton, S.; Dán, Á.; Bányai, K.; Fehér, E. Occurrence and Genetic Diversity of CRESS DNA Viruses in Wild Birds: A Hungarian Study. Sci. Rep. 2020, 10, 7036. [Google Scholar] [CrossRef]
- Liu, T.-N.; Liu, C.-X.; Liao, J.-Y.; Xiong, W.-J.; Xia, J.-Y.; Xiao, C.-T. Identification and Genomic Characterization of a Novel Porcine CRESS DNA Virus from a Pig Suffering from Diarrhea in China. Arch. Virol. 2022, 167, 1355–1359. [Google Scholar] [CrossRef]
- Guo, Z.; He, Q.; Tang, C.; Zhang, B.; Yue, H. Identification and Genomic Characterization of a Novel CRESS DNA Virus from a Calf with Severe Hemorrhagic Enteritis in China. Virus Res. 2018, 255, 141–146. [Google Scholar] [CrossRef]
- Rosario, K.; Mettel, K.A.; Greco, A.M.; Breitbart, M. Prevalence of a Vertically Transmitted Single-Stranded DNA Virus in Spinybacked Orbweavers (Gasteracantha cancriformis) from Florida, USA. J. Gen. Virol. 2019, 100, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Teng, J.L.L.; Tsang, A.K.L.; Joseph, M.; Wong, E.Y.M.; Tang, Y.; Sivakumar, S.; Bai, R.; Wernery, R.; et al. Metagenomic Analysis of Viromes of Dromedary Camel Fecal Samples Reveals Large Number and High Diversity of Circoviruses and Picobirnaviruses. Virology 2014, 471–473, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Orton, J.P.; Morales, M.; Fontenele, R.S.; Schmidlin, K.; Kraberger, S.; Leavitt, D.J.; Webster, T.H.; Wilson, M.A.; Kusumi, K.; Dolby, G.A.; et al. Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses. Viruses 2020, 12, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catoni, M.; Noris, E.; Vaira, A.M.; Jonesman, T.; Matić, S.; Soleimani, R.; Behjatnia, S.A.A.; Vinals, N.; Paszkowski, J.; Accotto, G.P. Virus-Mediated Export of Chromosomal DNA in Plants. Nat. Commun. 2018, 9, 5308. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus Taxonomy in the Age of Metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Viruses with Circular Single-Stranded DNA Genomes Are Everywhere!|Annual Review of Virology. Available online: https://www.annualreviews.org/doi/10.1146/annurev-virology-101416-041953 (accessed on 25 April 2022).
- Varsani, A.; Krupovic, M. Smacoviridae: A New Family of Animal-Associated Single-Stranded DNA Viruses. Arch. Virol. 2018, 163, 2005–2015. [Google Scholar] [CrossRef] [PubMed]
- Delwart, E.; Li, L. Rapidly Expanding Genetic Diversity and Host Range of the Circoviridae Viral Family and Other Rep Encoding Small Circular SsDNA Genomes. Virus Res. 2012, 164, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Phan, T.G.; da Costa, A.C.; del Valle Mendoza, J.; Bucardo-Rivera, F.; Nordgren, J.; O’Ryan, M.; Deng, X.; Delwart, E. The Fecal Virome of South and Central American Children with Diarrhea Includes Small Circular DNA Viral Genomes of Unknown Origin. Arch. Virol. 2016, 161, 959–966. [Google Scholar] [CrossRef]
- Klenner, J.; Kohl, C.; Dabrowski, P.W.; Nitsche, A. Comparing Viral Metagenomic Extraction Methods. Curr. Issues Mol. Biol. 2017, 24, 59–70. [Google Scholar] [CrossRef]
- Van Borm, S.; Fu, Q.; Winand, R.; Vanneste, K.; Hakhverdyan, M.; Höper, D.; Vandenbussche, F. Evaluation of a Commercial Exogenous Internal Process Control for Diagnostic RNA Virus Metagenomics from Different Animal Clinical Samples. J. Virol. Methods 2020, 283, 113916. [Google Scholar] [CrossRef]
- Siqueira, J.D.; Dominguez-Bello, M.G.; Contreras, M.; Lander, O.; Caballero-Arias, H.; Xutao, D.; Noya-Alarcon, O.; Delwart, E. Complex Virome in Feces from Amerindian Children in Isolated Amazonian Villages. Nat. Commun. 2018, 9, 4270. [Google Scholar] [CrossRef] [PubMed]
- Ensemble Strategy That Significantly Improves de Novo Assembly of Microbial Genomes from Metagenomic Next-Generation Sequencing Data|Nucleic Acids Research|Oxford Academic. Available online: https://academic.oup.com/nar/article/43/7/e46/2414255 (accessed on 25 April 2022).
- Okonechnikov, K.; Golosova, O.; Fursov, M. UGENE team Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinform. Oxf. Engl. 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid Annotation of Viral Genomes Using a Closely Related Reference Genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinformatics 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Evolutionary Trees from DNA Sequences: A Maximum Likelihood Approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Posada, D. JModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Do Socorro Fôro Ramos, E.; de Oliveira Ribeiro, G.; Villanova, F.; de Padua Milagres, F.A.; Brustulin, R.; Araújo, E.L.L.; Pandey, R.P.; Raj, V.S.; Deng, X.; Delwart, E.; et al. Composition of Eukaryotic Viruses and Bacteriophages in Individuals with Acute Gastroenteritis. Viruses 2021, 13, 2365. [Google Scholar] [CrossRef] [PubMed]
- Do Socorro Fôro Ramos, E.; Rosa, U.A.; de Oliveira Ribeiro, G.; Villanova, F.; de Pádua Milagres, F.A.; Brustulin, R.; Dos Santos Morais, V.; Bertanhe, M.; Marcatti, R.; Araújo, E.L.L.; et al. High Heterogeneity of Echoviruses in Brazilian Children with Acute Gastroenteritis. Viruses 2021, 13, 595. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Vanhulle, E.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; Shi, C.; Ghogomu, S.M.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Gut Virome Analysis of Cameroonians Reveals High Diversity of Enteric Viruses, Including Potential Interspecies Transmitted Viruses. mSphere 2019, 4, e00585-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Genbank ID (Genome Length in bp 1) | Host */Date/Location | SmaCV3BR08 | SmaCV3BR291 |
|---|---|---|---|
| Identity | |||
| NC026317 (2485) | Alouatta caraya/2009/USA | 0.874 | 0.884 |
| MG522850 (2511) | H. sapiens/2016/Ethiopia | 0.650 | 0.643 |
| KX838317 (2581) | H.sapiens/2012/Botswana | 0.640 | 0.643 |
| NC026318 (2529) | P. troglodytes/2012/USA | 0.660 | 0.654 |
| MH933796 (2538) | H.sapiens/2014/Cameroon | 0.650 | 0.651 |
| MF347417 (2560) | H.sapiens/?/Peru | 0.644 | 0.646 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villanova, F.; Milagres, F.A.d.P.; Brustulin, R.; Araújo, E.L.L.; Pandey, R.P.; Raj, V.S.; Deng, X.; Delwart, E.; Luchs, A.; Costa, A.C.d.; et al. A New Circular Single-Stranded DNA Virus Related with Howler Monkey Associated Porprismacovirus 1 Detected in Children with Acute Gastroenteritis. Viruses 2022, 14, 1472. https://doi.org/10.3390/v14071472
Villanova F, Milagres FAdP, Brustulin R, Araújo ELL, Pandey RP, Raj VS, Deng X, Delwart E, Luchs A, Costa ACd, et al. A New Circular Single-Stranded DNA Virus Related with Howler Monkey Associated Porprismacovirus 1 Detected in Children with Acute Gastroenteritis. Viruses. 2022; 14(7):1472. https://doi.org/10.3390/v14071472
Chicago/Turabian StyleVillanova, Fabiola, Flávio Augusto de Padua Milagres, Rafael Brustulin, Emerson Luiz Lima Araújo, Ramendra Pati Pandey, V. Samuel Raj, Xutao Deng, Eric Delwart, Adriana Luchs, Antonio Charlys da Costa, and et al. 2022. "A New Circular Single-Stranded DNA Virus Related with Howler Monkey Associated Porprismacovirus 1 Detected in Children with Acute Gastroenteritis" Viruses 14, no. 7: 1472. https://doi.org/10.3390/v14071472
APA StyleVillanova, F., Milagres, F. A. d. P., Brustulin, R., Araújo, E. L. L., Pandey, R. P., Raj, V. S., Deng, X., Delwart, E., Luchs, A., Costa, A. C. d., & Leal, É. (2022). A New Circular Single-Stranded DNA Virus Related with Howler Monkey Associated Porprismacovirus 1 Detected in Children with Acute Gastroenteritis. Viruses, 14(7), 1472. https://doi.org/10.3390/v14071472