
Expanded Diversity and Host Range of Bovine Hepacivirus—Genomic and Serological Evidence in Domestic and Wild Ruminant Species

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Serum Samples
2.2. Screening for Viral Genome Fragments in Domestic and Wild Ruminants
2.3. Next Generation Sequencing and Bioinformatic Analysis
2.4. Phylogenetic Analysis
2.5. Luciferase Immunoprecipitation System (LIPS) Assay
3. Results
3.1. Identification and Genetic Characterization of BovHepV in Bulgarian Cattle
3.2. Identification and Genetic Characterization of BovHepV in Wild Ruminants
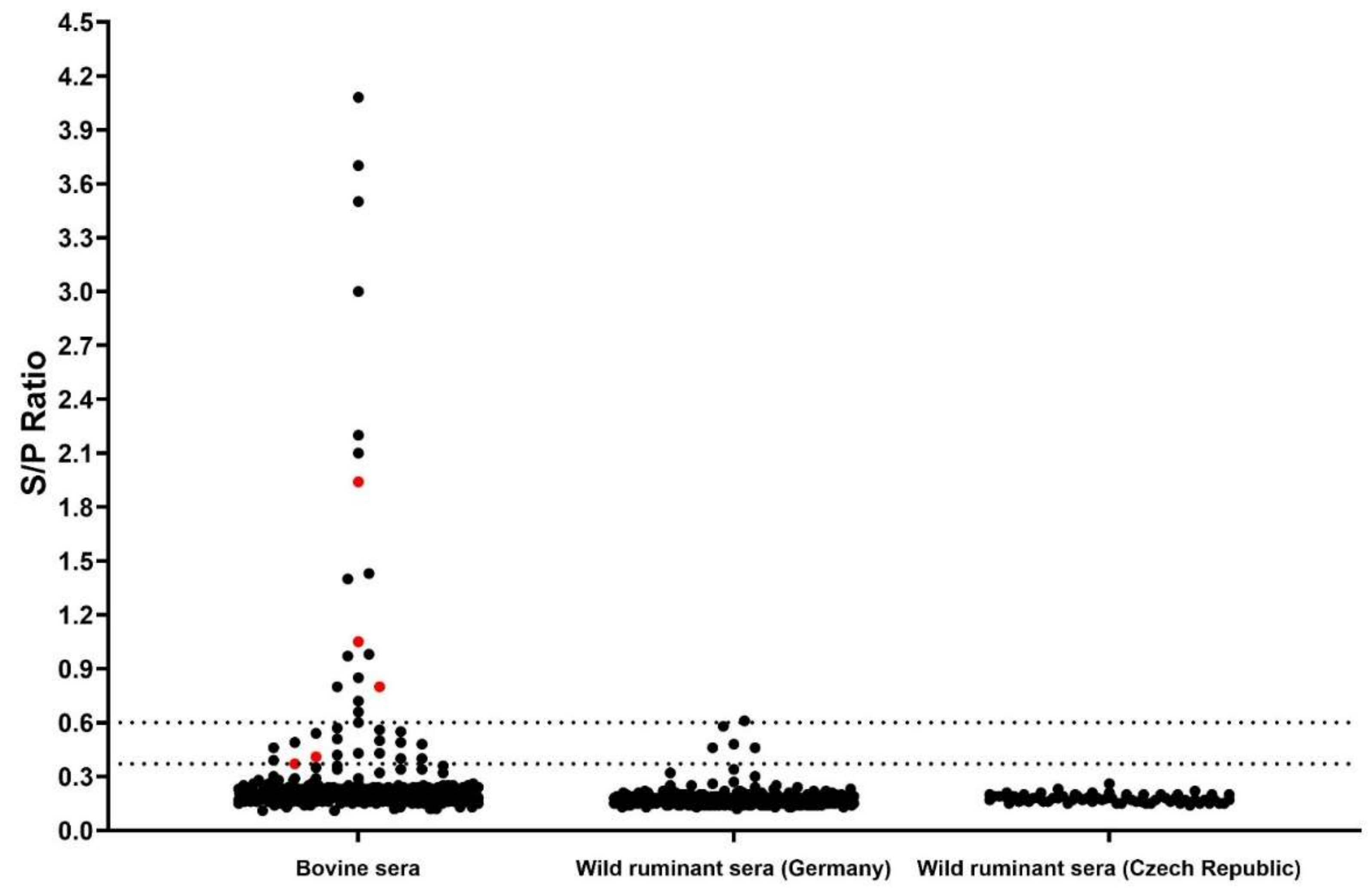
3.3. Luciferase Immunoprecipitation System (LIPS) Assay
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartenschlager, R.; Baumert, T.F.; Bukh, J.; Houghton, M.; Lemon, S.M.; Lindenbach, B.D.; Lohmann, V.; Moradpour, D.; Pietschmann, T.; Rice, C.M.; et al. Critical challenges and emerging opportunities in hepatitis C virus research in an era of potent antiviral therapy: Considerations for scientists and funding agencies. Virus Res. 2018, 248, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J. Animal models for the study of hepatitis C virus infection and related liver disease. Gastroenterology 2012, 142, 1279–1287.e3. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.L.; Schoeniger, A.; Becher, P.; Baechlein, C. Mutational analysis of the bovine hepacivirus internal ribosome entry site. J. Virol. 2018, 92, e01974-17. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV virus taxonomy profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef]
- Kapoor, A.; Simmonds, P.; Gerold, G.; Qaisar, N.; Jain, K.; Henriquez, J.A.; Firth, C.; Hirschberg, D.L.; Rice, C.M.; Shields, S.; et al. Characterization of a canine homolog of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 11608–11613. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Dubovi, E.J.; Simmonds, P.; Medina, J.L.; Henriquez, J.A.; Mishra, N.; Wagner, J.; Tokarz, R.; Cullen, J.M.; Iadarola, M.J.; et al. Serology-enabled discovery of genetically diverse hepaciviruses in a new host. J. Virol. 2012, 86, 6171–6178. [Google Scholar] [CrossRef]
- Smith, D.B.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, A.S.; Pletnev, A.; Rico-Hesse, R.; Stapleton, J.T.; et al. Proposed update to the taxonomy of the genera hepacivirus and pegivirus within the flaviviridae family. J. Gen. Virol. 2016, 97, 2894–2907. [Google Scholar] [CrossRef]
- Lauck, M.; Sibley, S.D.; Lara, J.; Purdy, M.A.; Khudyakov, Y.; Hyeroba, D.; Tumukunde, A.; Weny, G.; Switzer, W.M.; Chapman, C.A.; et al. A novel hepacivirus with an unusually long and intrinsically disordered NS5A protein in a wild old world primate. J. Virol. 2013, 87, 8971–8981. [Google Scholar] [CrossRef]
- Kapoor, A.; Simmonds, P.; Scheel, T.K.H.; Hjelle, B.; Cullen, J.M.; Burbelo, P.D.; Chauhan, L.V.; Duraisamy, R.; Sanchez Leon, M.; Jain, K.; et al. Identification of rodent homologs of hepatitis C virus and pegiviruses. mBio 2013, 4, e00216-13. [Google Scholar] [CrossRef]
- Quan, P.; Firth, C.; Conte, J.M.; Williams, S.H.; Zambrana-Torrelio, C.; Anthony, S.; Ellison, J.; Gilbert, A.; Kuzmin, I.; Niezgoda, M.; et al. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc. Natl. Acad. Sci. USA 2013, 110, 8194–8199. [Google Scholar] [CrossRef] [PubMed]
- Baechlein, C.; Fischer, N.; Grundhoff, A.; Alawi, M.; Indenbirken, D.; Postel, A.; Baron, A.L.; Offinger, J.; Becker, K.; Beineke, A.; et al. Identification of a novel hepacivirus in domestic cattle from Germany. J. Virol. 2015, 89, 7007–7015. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.; Grundhoff, A.; Baechlein, C.; Fischer, N.; Gmyl, A.; Wollny, R.; Dei, D.; Ritz, D.; Binger, T.; Adankwah, E.; et al. Highly divergent hepaciviruses from African cattle. J. Virol. 2015, 89, 5876–5882. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, L.; Liu, M.; Shen, S.; Zhang, Y.; Xu, Y.; Deng, H.; Deng, F.; Duan, Z. Detection and characterization of a novel hepacivirus in long-tailed ground squirrels (spermophilus undulatus) in China. Arch. Virol. 2019, 164, 2401–2410. [Google Scholar] [CrossRef]
- Chang, W.-S.; Eden, J.-S.; Hartley, W.J.; Shi, M.; Rose, K.; Holmes, E.C. Metagenomic discovery and co-infection of diverse wobbly possum disease viruses and a novel hepacivirus in Australian brushtail possums. One Health Outlook 2019, 1, 5. [Google Scholar] [CrossRef]
- Moreira-Soto, A.; Arroyo-Murillo, F.; Sander, A.-L.; Rasche, A.; Corman, V.; Tegtmeyer, B.; Steinmann, E.; Corrales-Aguilar, E.; Wieseke, N.; Avey-Arroyo, J.; et al. Cross-order host switches of hepatitis c-related viruses illustrated by a novel hepacivirus from sloths. Virus Evol. 2020, 6, veaa033. [Google Scholar] [CrossRef]
- Guo, H.; Cai, C.; Wang, B.; Zhuo, F.; Jiang, R.; Wang, N.; Li, B.; Zhang, W.; Zhu, Y.; Fan, Y.; et al. Novel hepacivirus in Asian house shrew, China. Sci. China Life Sci. 2019, 62, 701–704. [Google Scholar] [CrossRef]
- Porter, A.F.; Pettersson, J.H.-O.; Chang, W.-S.; Harvey, E.; Rose, K.; Shi, M.; Eden, J.-S.; Buchmann, J.; Moritz, C.; Holmes, E.C. Novel hepaci- and pegi-like viruses in native Australian wildlife and non-human primates. Virus Evol. 2020, 6, veaa064. [Google Scholar] [CrossRef]
- Chu, L.-S.; Jin, M.; Feng, C.; Wang, X.; Zhang, D. A highly divergent hepacivirus-like flavivirus in domestic ducks. J. Gen. Virol. 2019, 100, 1234–1240. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Vasilakis, N.; Tian, J.-H.; Li, C.-X.; Chen, L.-J.; Eastwood, G.; Diao, X.-N.; Chen, M.-H.; Chen, X.; et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the flaviviridae and related viruses. J. Virol. 2015, 90, 659–669. [Google Scholar] [CrossRef]
- Harvey, E.; Rose, K.; Eden, J.-S.; Lo, N.; Abeyasuriya, T.; Shi, M.; Doggett, S.L.; Holmes, E.C. Extensive diversity of RNA viruses in Australian ticks. J. Virol. 2019, 93, e01358-18. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.H.; Levy, A.; Yates, R.A.; Somaweera, N.; Neville, P.J.; Nicholson, J.; Lindsay, M.D.A.; Mackenzie, J.S.; Jain, K.; Imrie, A.; et al. Discovery of Jogalong virus, a novel hepacivirus identified in a Culex annulirostris (Skuse) mosquito from the Kimberley region of Western Australia. PLoS ONE 2020, 15, e0227114. [Google Scholar] [CrossRef]
- Wolfisberg, R.; Holmbeck, K.; Nielsen, L.; Kapoor, A.; Rice, C.M.; Bukh, J.; Scheel, T.K.H. Replicons of a rodent hepatitis C model virus permit selection of highly permissive cells. J. Virol. 2019, 93, e00733-19. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Lukashev, A.N.; Gmyl, A.; Coutard, B.; Adam, A.; Ritz, D.; Leijten, L.M.; van Riel, D.; et al. Evidence for novel hepaciviruses in rodents. PLoS Pathog. 2013, 9, e1003438. [Google Scholar] [CrossRef]
- Canuti, M.; Williams, C.V.; Sagan, S.M.; Oude Munnink, B.B.; Gadi, S.; Verhoeven, J.T.P.; Kellam, P.; Cotten, M.; Lang, A.S.; Junge, R.E.; et al. Virus discovery reveals frequent infection by diverse novel members of the flaviviridae in wild lemurs. Arch. Virol. 2019, 164, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Rasche, A.; Moreira-Soto, A.; Pfaender, S.; Bletsa, M.; Corman, V.M.; Aguilar-Setien, A.; García-Lacy, F.; Hans, A.; Todt, D.; et al. Differential infection patterns and recent evolutionary origins of equine hepaciviruses in donkeys. J. Virol. 2017, 91, e01711-16. [Google Scholar] [CrossRef]
- Canal, C.; Weber, M.N.; Cibulski, S.; Silva, M.S.; Puhl, D.E.; Stalder, H.; Peterhans, E. A novel genetic group of bovine hepacivirus in archival serum samples from Brazilian cattle. BioMed Res. Int. 2017, 2017, 4732520. [Google Scholar] [CrossRef]
- Elia, G.; Caringella, F.; Lanave, G.; Martella, V.; Losurdo, M.; Tittarelli, M.; Colitti, B.; Decaro, N.; Buonavoglia, C. Genetic heterogeneity of bovine hepacivirus in Italy. Transbound. Emerg. Dis. 2020, 67, 2731–2740. [Google Scholar] [CrossRef]
- Sadeghi, M.; Kapusinszky, B.; Yugo, D.M.; Phan, T.G.; Deng, X.; Kanevsky, I.; Opriessnig, T.; Woolums, A.R.; Hurley, D.J.; Meng, X.-J.; et al. Virome of US calf serum. Biologicals 2017, 46, 64–67. [Google Scholar] [CrossRef]
- Lu, G.; Jia, K.; Ping, X.; Huang, J.; Luo, A.; Wu, P.; Li, S. Novel bovine hepacivirus in dairy cattle, China. Emerg. Microbes Infect. 2018, 7, 54. [Google Scholar] [CrossRef]
- Yeşilbağ, K.; Baechlein, C.; Kadiroğlu, B.; Baldan Toker, E.; Alpay, G.; Becher, P. Presence of bovine hepacivirus in Turkish cattle. Vet. Microbiol. 2018, 225, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Baechlein, C.; Baron, A.L.; Meyer, D.; Gorriz-Martin, L.; Pfankuche, V.M.; Baumgärtner, W.; Polywka, S.; Peine, S.; Fischer, N.; Rehage, J.; et al. Further characterization of bovine hepacivirus: Antibody response, course of infection, and host tropism. Transbound. Emerg. Dis. 2019, 66, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Pfaender, S.; Cavalleri, J.M.V.; Walter, S.; Doerrbecker, J.; Campana, B.; Brown, R.J.P.; Burbelo, P.D.; Postel, A.; Hahn, K.; Anggakusuma; et al. Clinical course of infection and viral tissue tropism of hepatitis C virus-like nonprimate hepaciviruses in horses. Hepatology 2015, 61, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.; Shen, X.; Peng, H.; Guo, X.; He, Z.; Yao, M.; Fu, G.; Cui, Y.; Zhang, X.; Huang, Y.; et al. Complete genome sequence of a novel bovine hepacivirus from Yunnan, China. Arch. Virol. 2020, 165, 1489–1494. [Google Scholar] [CrossRef]
- Lu, G.; Ou, J.; Zhao, J.; Li, S. Presence of a novel subtype of bovine hepacivirus in China and expanded classification of bovine hepacivirus strains worldwide into 7 subtypes. Viruses 2019, 11, 843. [Google Scholar] [CrossRef]
- Shao, J.-W.; Guo, L.-Y.; Yuan, Y.-X.; Ma, J.; Chen, J.-M.; Liu, Q. A novel subtype of bovine hepacivirus identified in ticks reveals the genetic diversity and evolution of bovine hepacivirus. Viruses 2021, 13, 2206. [Google Scholar] [CrossRef]
- Schlottau, K.; Wernike, K.; Forth, L.; Holsteg, M.; Höper, D.; Beer, M.; Hoffmann, B. Presence of two different bovine hepacivirus clusters in Germany. Transbound. Emerg. Dis. 2018, 65, 1705–1711. [Google Scholar] [CrossRef]
- Da Silva, M.S.; Junqueira, D.M.; Baumbach, L.F.; Cibulski, S.P.; Mósena, A.C.S.; Weber, M.N.; Silveira, S.; de Moraes, G.M.; Maia, R.D.; Coimbra, V.C.S.; et al. Comprehensive evolutionary and phylogenetic analysis of hepacivirus N (HNV). J. Gen. Virol. 2018, 99, 890–896. [Google Scholar] [CrossRef]
- Da Silva, M.S.; Weber, M.N.; Baumbach, L.F.; Cibulski, S.P.; Budaszewski, R.F.; Mósena, A.C.S.; Canova, R.; Varela, A.P.M.; Mayer, F.Q.; Canal, C.W. Highly divergent cattle hepacivirus N in Southern Brazil. Arch. Virol. 2019, 164, 3133–3136. [Google Scholar] [CrossRef]
- Alawi, M.; Burkhardt, L.; Indenbirken, D.; Reumann, K.; Christopeit, M.; Kröger, N.; Lütgehetmann, M.; Aepfelbacher, M.; Fischer, N.; Grundhoff, A. DAMIAN: An open source bioinformatics tool for fast, systematic and cohort based analysis of microorganisms in diagnostic samples. Sci. Rep. 2019, 9, 16841. [Google Scholar] [CrossRef]
- Hall, T. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for WINDOWS 95/98/NT; Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; Volume 41. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Thézé, J.; Lowes, S.; Parker, J.; Pybus, O.G. Evolutionary and phylogenetic analysis of the hepaciviruses and pegiviruses. Genome Biol. Evol. 2015, 7, 2996–3008. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soza, A.; Riquelme, A.; Arrese, M. Routes of transmission of hepatitis C virus. Ann. Hepatol. 2010, 9 (Suppl. 1), S30–S33. [Google Scholar] [CrossRef]
- Ramsay, J.D.; Evanoff, R.; Wilkinson, T.E., Jr.; Divers, T.J.; Knowles, D.P.; Mealey, R.H. Experimental transmission of equine hepacivirus in horses as a model for hepatitis C virus. Hepatology 2015, 61, 1533–1546. [Google Scholar] [CrossRef]
- Tovo, P.-A.; Calitri, C.; Scolfaro, C.; Gabiano, C.; Garazzino, S. Vertically acquired hepatitis C virus infection: Correlates of transmission and disease progression. World J. Gastroenterol. 2016, 22, 1382–1392. [Google Scholar] [CrossRef]
- Gather, T.; Walter, S.; Todt, D.; Pfaender, S.; Brown, R.J.P.; Postel, A.; Becher, P.; Moritz, A.; Hansmann, F.; Baumgaertner, W.; et al. Vertical transmission of hepatitis C virus-like non-primate hepacivirus in horses. J. Gen. Virol. 2016, 97, 2540–2551. [Google Scholar] [CrossRef]
- Pronost, S.; Fortier, C.; Marcillaud-Pitel, C.; Tapprest, J.; Foursin, M.; Saunier, B.; Pitel, P.-H.; Paillot, R.; Hue, E.S. Further evidence for in utero transmission of equine hepacivirus to foals. Viruses 2019, 11, 1124. [Google Scholar] [CrossRef]
- Done, J.T.; Terlecki, S.; Richardson, C.; Harkness, J.W.; Sands, J.J.; Patterson, D.S.; Sweasey, D.; Shaw, I.G.; Winkler, C.E.; Duffell, S.J. Bovine virus diarrhoea-mucosal disease virus: Pathogenicity for the fetal calf following maternal infection. Vet. Rec. 1980, 106, 473–479. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Kovacs, J.A.; Ching, K.H.; Issa, A.T.; Iadarola, M.J.; Murphy, A.A.; Schlaak, J.F.; Masur, H.; Polis, M.A.; Kottilil, S. Proteome-wide anti-HCV and anti-HIV antibody profiling for predicting and monitoring response to HCV treatment in HIV co-infected patients. J. Infect. Dis. 2010, 202, 894–898. [Google Scholar] [CrossRef]
- Tanaka, T.; Kasai, H.; Yamashita, A.; Okuyama-Dobashi, K.; Yasumoto, J.; Maekawa, S.; Enomoto, N.; Okamoto, T.; Matsuura, Y.; Morimatsu, M.; et al. Hallmarks of hepatitis C virus in equine hepacivirus. J. Virol. 2014, 88, 13352–13366. [Google Scholar] [CrossRef]
- Lyons, S.; Kapoor, A.; Schneider, B.S.; Wolfe, N.D.; Culshaw, G.; Corcoran, B.; Durham, A.E.; Burden, F.; McGorum, B.C.; Simmonds, P. Viraemic frequencies and seroprevalence of non-primate hepacivirus and equine pegiviruses in horses and other mammalian species. J. Gen. Virol. 2014, 95, 1701–1711. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Badenhorst, M.; Tegtmeyer, B.; Todt, D.; Guthrie, A.; Feige, K.; Campe, A.; Steinmann, E.; Cavalleri, J.M.V. First detection and frequent occurrence of equine hepacivirus in horses on the african continent. Vet. Microbiol. 2018, 223, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Reichert, C.; Campe, A.; Walter, S.; Pfaender, S.; Welsch, K.; Ruddat, I.; Sieme, H.; Feige, K.; Steinmann, E.; Cavalleri, J.M.V. Frequent occurrence of nonprimate hepacivirus infections in thoroughbred breeding horses—A cross-sectional study for the occurrence of infections and potential risk factors. Vet. Microbiol. 2017, 203, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Matsuu, A.; Hobo, S.; Ando, K.; Sanekata, T.; Sato, F.; Endo, Y.; Amaya, T.; Osaki, T.; Horie, M.; Masatani, T.; et al. Genetic and serological surveillance for non-primate hepacivirus in horses in Japan. Vet. Microbiol. 2015, 179, 219–227. [Google Scholar] [CrossRef]
- Badenhorst, M.; de Heus, P.; Auer, A.; Rümenapf, T.; Tegtmeyer, B.; Kolodziejek, J.; Nowotny, N.; Steinmann, E.; Cavalleri, J.-M.V. No evidence of mosquito involvement in the transmission of equine hepacivirus (flaviviridae) in an epidemiological survey of Austrian horses. Viruses 2019, 11, 1014. [Google Scholar] [CrossRef]
- Tomlinson, J.; Wolfisberg, R.; Fahnøe, U.; Patel, R.; Trivedi, S.; Kumar, A.; Sharma, H.; Nielsen, L.; McDonough, S.; Bukh, J.; et al. Pathogenesis, MiR-122 gene-regulation, and protective immune responses after acute equine hepacivirus infection. Hepatology 2021, 74, 1148–1163. [Google Scholar] [CrossRef] [PubMed]
- El-Attar, L.M.R.; Mitchell, J.A.; Brooks Brownlie, H.; Priestnall, S.L.; Brownlie, J. Detection of non-primate hepaciviruses in UK dogs. Virology 2015, 484, 93–102. [Google Scholar] [CrossRef]
- Abbadi, I.; Lkhider, M.; Kitab, B.; Jabboua, K.; Zaidane, I.; Haddaji, A.; Nacer, S.; Matsuu, A.; Pineau, P.; Tsukiyama-Kohara, K.; et al. Non-primate hepacivirus transmission and prevalence: Novel findings of virus circulation in horses and dogs in Morocco. Infect. Genet. Evol. 2021, 93, 104975. [Google Scholar] [CrossRef]
- Hartlage, A.S.; Cullen, J.M.; Kapoor, A. The strange, expanding world of animal hepaciviruses. Annu. Rev. Virol. 2016, 3, 53–75. [Google Scholar] [CrossRef]
- Anggakusuma; Brown, R.J.P.; Banda, D.H.; Todt, D.; Vieyres, G.; Steinmann, E.; Pietschmann, T. Hepacivirus NS3/4A proteases interfere with MAVS signaling in both their cognate animal hosts and humans: Implications for zoonotic transmission. J. Virol. 2016, 90, 10670–10681. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer/Probe | PCR Assay | Animal Species | Sequence (5′-3′) | Source |
|---|---|---|---|---|
| BovHepV_5NTR_fwd | RT-qPCR | Bovine | AACAGGCCCCTAGTAG | Baechlein et al. [32] |
| BovHepV_5NTR_rev | RT-qPCR | Bovine | GTACTCGGTCCTTCCCA | Baechlein et al. [32] |
| BovHepV_5NTR_probe | RT-qPCR | Bovine | CATGAGCCCTTTCCCCACAGATTGAGTGGA | Baechlein et al. [32] |
| Pan-hepaci-NS3_fwd | Nested RT-PCR | Bovine | GCMCCTACKGGSTCYGGGAA | Baechlein et al. [32] |
| Pan-hepaci-NS3_rev | Nested RT-PCR | Bovine | TCRAAGTTCCCRGTGTAMCCMGTCAT | Baechlein et al. [32] |
| Pan-hepaci-NS3_nested_fwd | Nested RT-PCR | Bovine | GAYGTGRTCATYTGTGATGARTGCCA | Baechlein et al. [32] |
| Pan-hepaci-NS3_nested_rev | Nested RT-PCR | Bovine | CCSCGATAGTARGCSACWGC | Baechlein et al. [32] |
| BovHepV_3511_fwd | RT-PCR | Bovine | TGGGARGTCCARACTGTCTATG | Baechlein et al. [32] |
| BovHepV_4608_rev | RT-PCR | Bovine | CTCATAACARATCTGCCTCTGC | Baechlein et al. [32] |
| CZ178_semi-nested_fwd_1 | Semi-nested RT-PCR | Wild ruminants | ATGTCGGCGATCTCAACTTCC | This study |
| CZ178_semi-nested_fwd_2 | Semi-nested RT-PCR | Wild ruminants | ACACTGTGAGAGTCTCGCAGC | This study |
| CZ178_semi-nested_rev_1 | Semi-nested RT-PCR | Wild ruminants | TGAGCACGAACGACGCTGTTG | This study |
| CZ178_semi-nested_fwd_3 | Semi-nested RT-PCR | Wild ruminants | CATGGTGCARCGGTGCAG | This study |
| CZ178_semi-nested_fwd_4 | Semi-nested RT-PCR | Wild ruminants | ATGCAYTATGTCCGAAAGGGC | This study |
| CZ178_semi-nested_rev_2 | Semi-nested RT-PCR | Wild ruminants | AGTAGCTGTGGCAAGCAGAAC | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breitfeld, J.; Fischer, N.; Tsachev, I.; Marutsov, P.; Baymakova, M.; Plhal, R.; Keuling, O.; Becher, P.; Baechlein, C. Expanded Diversity and Host Range of Bovine Hepacivirus—Genomic and Serological Evidence in Domestic and Wild Ruminant Species. Viruses 2022, 14, 1457. https://doi.org/10.3390/v14071457
Breitfeld J, Fischer N, Tsachev I, Marutsov P, Baymakova M, Plhal R, Keuling O, Becher P, Baechlein C. Expanded Diversity and Host Range of Bovine Hepacivirus—Genomic and Serological Evidence in Domestic and Wild Ruminant Species. Viruses. 2022; 14(7):1457. https://doi.org/10.3390/v14071457
Chicago/Turabian StyleBreitfeld, Jana, Nicole Fischer, Ilia Tsachev, Plamen Marutsov, Magdalena Baymakova, Radim Plhal, Oliver Keuling, Paul Becher, and Christine Baechlein. 2022. "Expanded Diversity and Host Range of Bovine Hepacivirus—Genomic and Serological Evidence in Domestic and Wild Ruminant Species" Viruses 14, no. 7: 1457. https://doi.org/10.3390/v14071457
APA StyleBreitfeld, J., Fischer, N., Tsachev, I., Marutsov, P., Baymakova, M., Plhal, R., Keuling, O., Becher, P., & Baechlein, C. (2022). Expanded Diversity and Host Range of Bovine Hepacivirus—Genomic and Serological Evidence in Domestic and Wild Ruminant Species. Viruses, 14(7), 1457. https://doi.org/10.3390/v14071457