
Development of Parallel Reaction Monitoring Mass Spectrometry Assay for the Detection of Human Norovirus Major Capsid Protein


Abstract
:1. Introduction
2. Materials and Method
2.1. HuNoV VP1 Protein Synthesis and Sample Preparation for MS Analysis
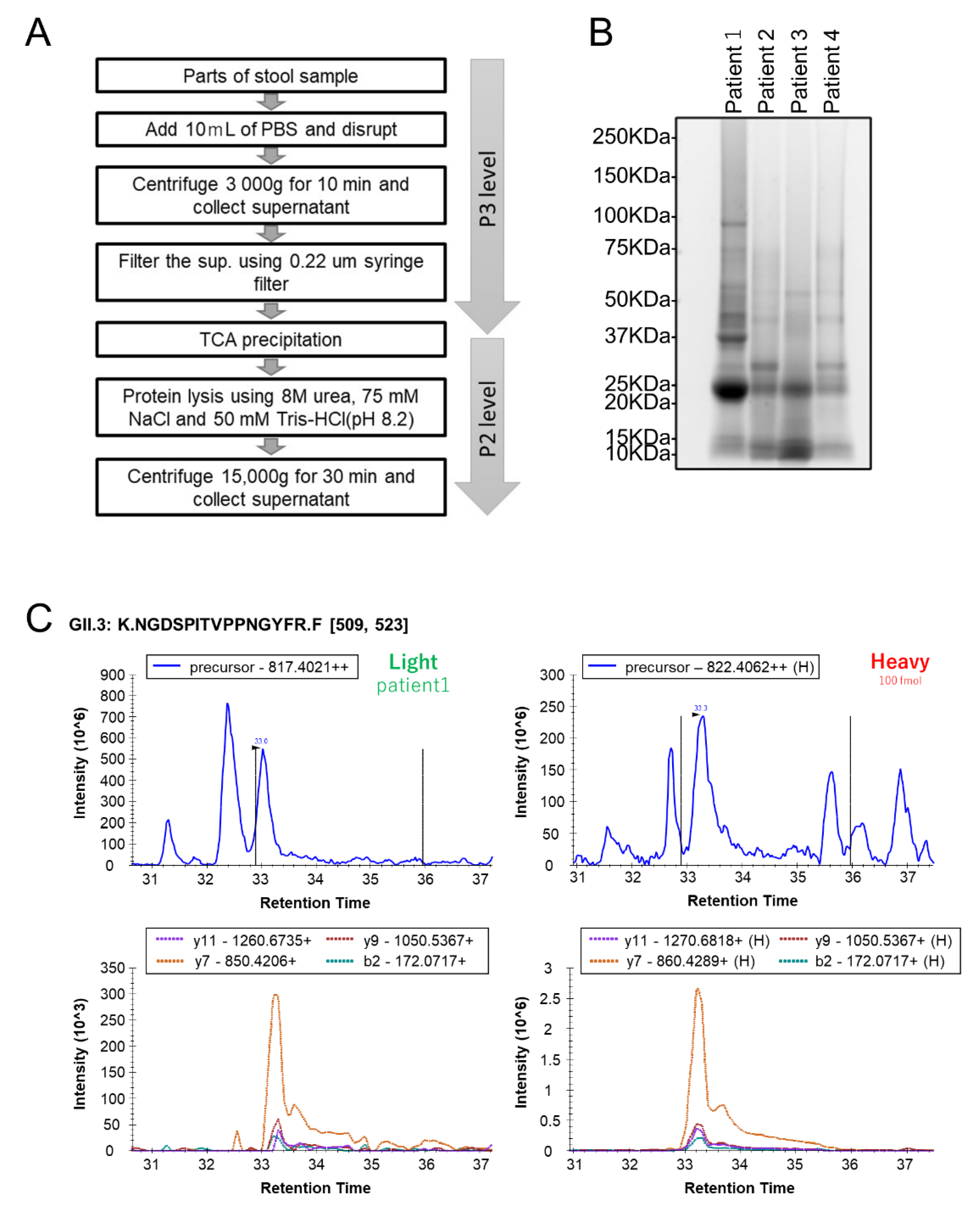
2.2. Human Stool Specimen Preparation for MS Analysis
2.3. Liquid Chromatography–Tandem Mass Spectrometry Analysis
2.4. Data Analysis
3. Results
3.1. Determination of PRM Analysis Target Peptides Using Recombinant Human Norovirus VP1
3.2. Optimization of PRM Assay Workflow and Quantification of VP1 Protein
3.3. Quantification of HuNoV VP1 by the Developed PRM Assays in Individual Human Stool Specimens
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de Graaf, M.; van Beek, J.; Koopmans, M.P. Human norovirus transmission and evolution in a changing world. Nat. Rev. Microbiol. 2016, 14, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Hall, A.J.; Vinje, J.; Parashar, U.D. Noroviruses: A comprehensive review. J. Clin. Virol. 2009, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, M.; Graham, D.Y.; Estes, M.K. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 1992, 66, 6527–6532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, J.N.; Graham, D.Y.; Wang, K.N.; Estes, M.K. Norwalk virus genome cloning and characterization. Science 1990, 250, 1580–1583. [Google Scholar]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef]
- Prasad, B.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray crystallographic structure of the Norwalk virus capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef] [Green Version]
- Caul, E.O.; Appleton, H. The electron microscopical and physical characteristics of small round human fecal viruses: An interim scheme for classification. J. Med. Virol. 1982, 9, 257–265. [Google Scholar] [CrossRef]
- Atmar, R.L.; Estes, M.K. Diagnosis of noncultivatable gastroenteritis viruses, the human caliciviruses. Clin. Microbiol. Rev. 2001, 14, 15–37. [Google Scholar] [CrossRef] [Green Version]
- Vinje, J. Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, T.; Kojima, S.; Shinohara, M.; Uchida, K.; Fukushi, S.; Hoshino, F.B.; Takeda, N.; Katayama, K. Broadly reactive and highly sensitive assay for Norwalk-like viruses based on real-time quantitative reverse transcription-PCR. J. Clin. Microbiol. 2003, 41, 1548–1557. [Google Scholar] [CrossRef] [Green Version]
- Rabenau, H.F.; Sturmer, M.; Buxbaum, S.; Walczok, A.; Preiser, W.; Doerr, H.W. Laboratory diagnosis of norovirus: Which method is the best? Intervirology 2003, 46, 232–238. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, E.; Duizer, E.; Vennema, H.; Koopmans, M.P. Diagnosis of Norovirus outbreaks by commercial ELISA or RT-PCR. J. Virol. Methods 2006, 137, 259–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doerflinger, S.Y.; Tabatabai, J.; Schnitzler, P.; Farah, C.; Rameil, S.; Sander, P.; Koromyslova, A.; Hansman, G.S. Development of a Nanobody-Based Lateral Flow Immunoassay for Detection of Human Norovirus. mSphere 2016, 1, e00219-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirato, H. ELISA-Based Methods to Detect and Quantify Norovirus Virus-Like Particle Attachment to Histo-Blood Group Antigens. Methods Mol. Biol. 2020, 2132, 597–607. [Google Scholar] [PubMed]
- Gallien, S.; Duriez, E.; Crone, C.; Kellmann, M.; Moehring, T.; Domon, B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol. Cell Proteom. 2012, 11, 1709–1723. [Google Scholar] [CrossRef] [Green Version]
- Bourmaud, A.; Gallien, S.; Domon, B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Principle and applications. Proteomics 2016, 16, 2146–2159. [Google Scholar] [CrossRef]
- Sawasaki, T.; Kamura, N.; Matsunaga, S.; Saeki, M.; Tsuchimochi, M.; Morishita, R.; Endo, Y. Arabidopsis HY5 protein functions as a DNA-binding tag for purification and functional immobilization of proteins on agarose/DNA microplate. FEBS Lett. 2008, 582, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Takahashi, C.; Moreland, N.J.; Chang, Y.T.; Sawasaki, T.; Ryo, A.; Vasudevan, S.G.; Suzuki, Y.; Yamamoto, N. Establishment of a robust dengue virus NS3-NS5 binding assay for identification of protein-protein interaction inhibitors. Antivir. Res. 2012, 96, 305–314. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Motoya, T.; Umezawa, M.; Saito, A.; Goto, K.; Doi, I.; Fukaya, S.; Nagata, N.; Ikeda, Y.; Okayama, K.; Aso, J.; et al. Variation of human norovirus GII genotypes detected in Ibaraki, Japan, during 2012–2018. Gut Pathog. 2019, 11, 26. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
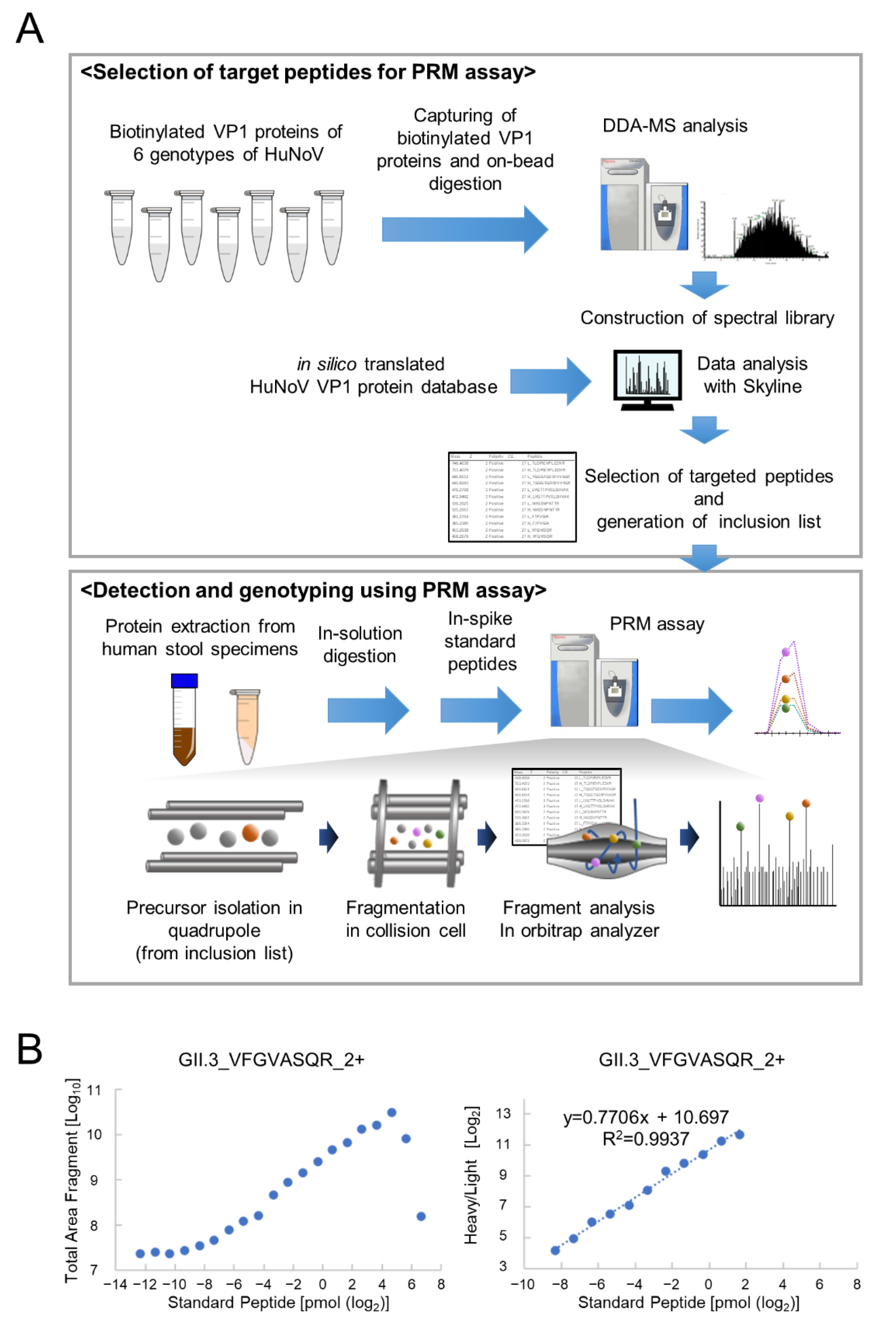

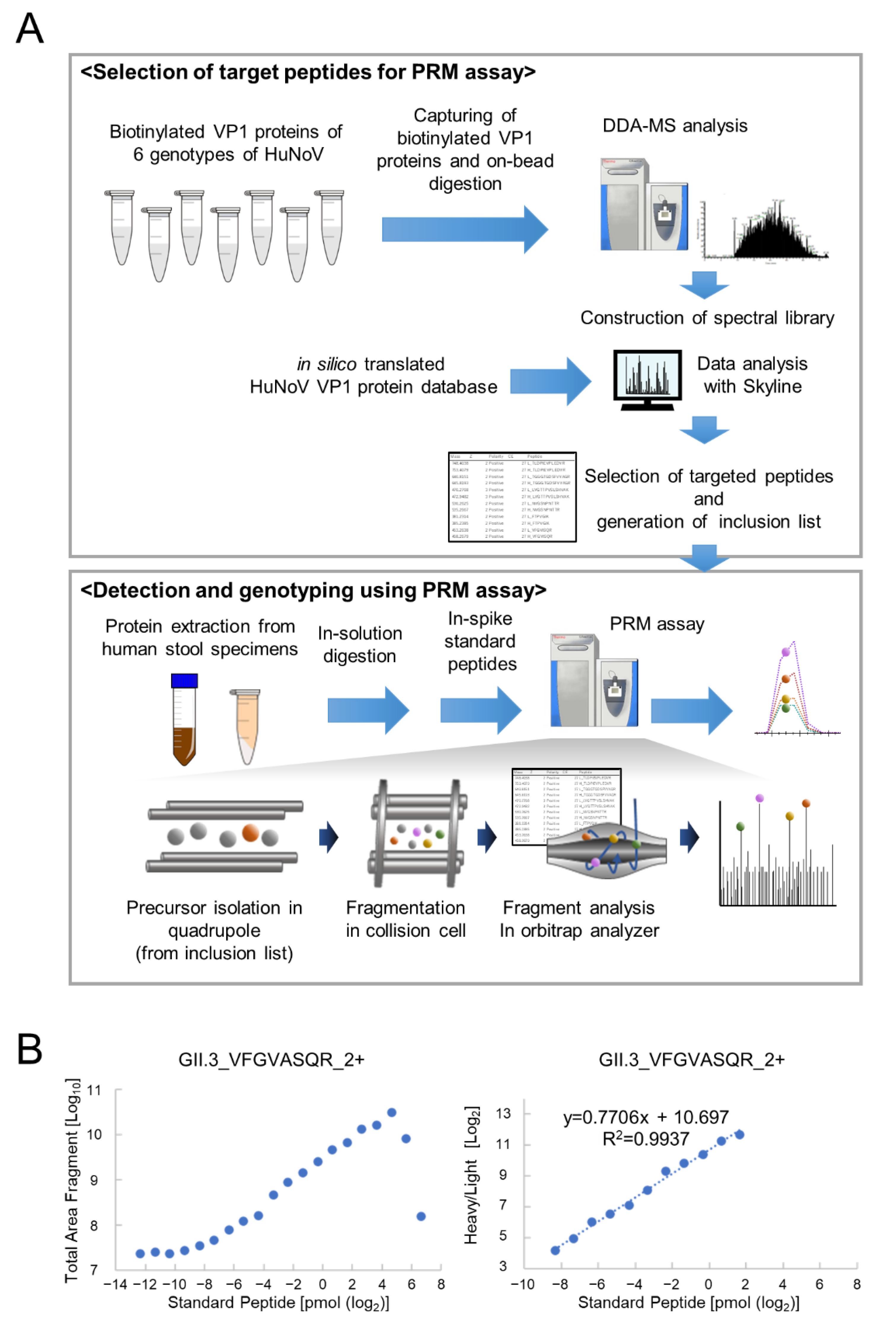{kind=link}
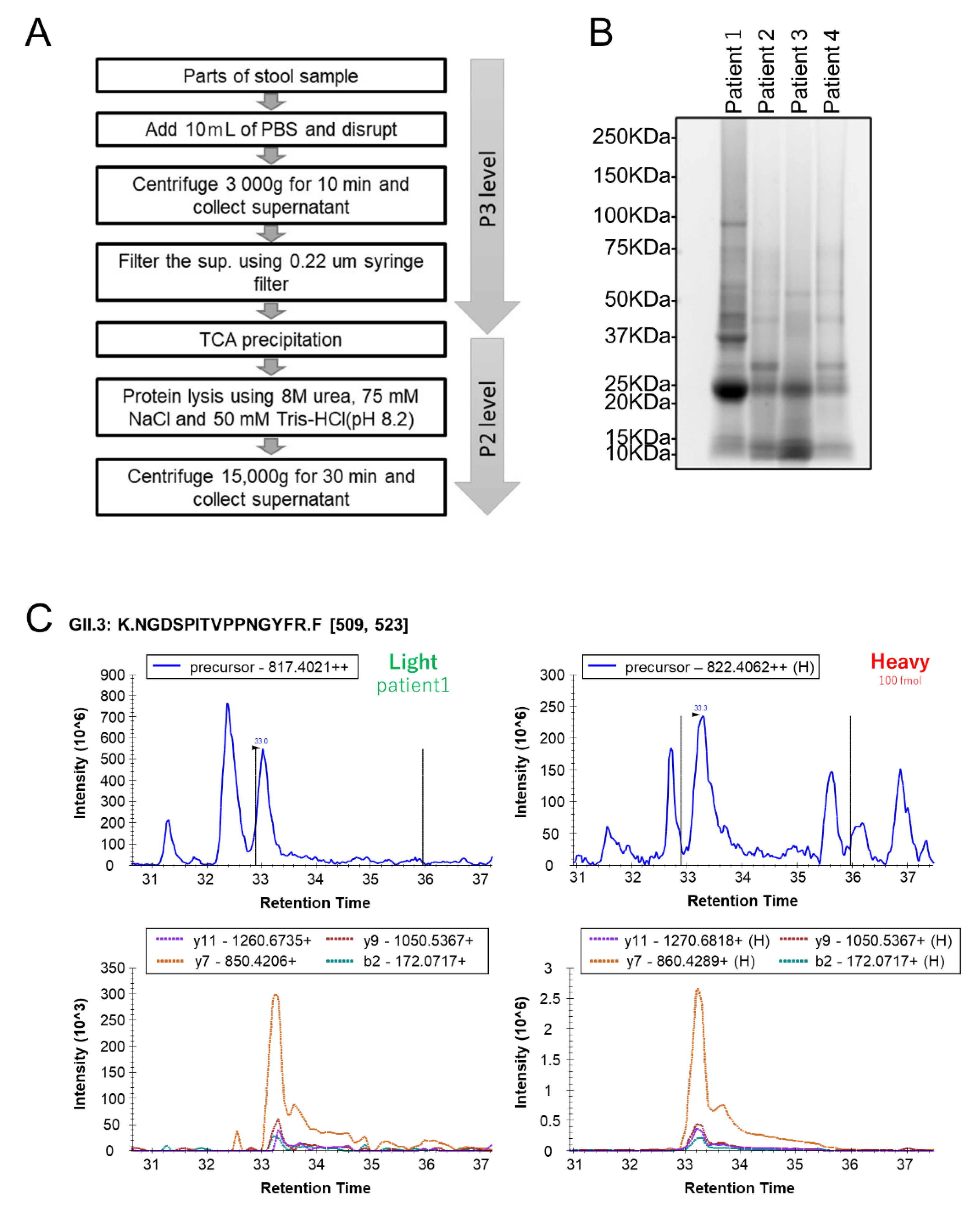{kind=link}
| Norovirus VP1 | Peptide | Start-End | Charge | Light m/z | Heavy m/z | Fragment Ions Analyzed | LLOD * (fmol) | LLOQ ** (fmol) | ULOQ *** (pmol) | CV of Light Peptide in the Linearity Range (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| GI.1_M87661/ Norwalk/68/US | TLDPIEVPLEDVR | 154–166 | 2 | 748.404 | 753.408 | y10+, y8+, y6+, y2+ | <0.19 | 780 | >100 | 23.19 |
| TGGGTGDSFVVAGR | 192–205 | 2 | 640.815 | 645.819 | y9+, y7+, y4+, y3+ | <0.19 | 3.05 | 25 | 23.92 | |
| LVGTTPVSLSHVAK | 276–289 | 3 | 470.277 | 472.948 | y9+, y8+, y7+, y2+ | <0.19 | 6.1 | 12.5 | 23.09 | |
| GII.2_X81879/ Melksham | VFGVISQR | 332–339 | 2 | 453.264 | 458.268 | y6+, y4+, y3+, b3+ | <0.19 | 3.05 | 12.5 | 23.10 |
| YAGALNLNTNLAPSVAPVFPGER | 410–432 | 2 | 1186.124 | 1191.128 | y12+, y11+, y7+, y4+ | <0.19 | 780 | 12.5 | 6.97 | |
| GII.3_U02030/ Tronto | VFGVASQR | 343–340 | 2 | 432.240 | 437.244 | y6+, y4+, b2+, b3+ | <0.19 | 3.05 | 3.13 | 23.15 |
| SQLPSSGGR | 443–451 | 2 | 444.730 | 449.734 | y7+, y6+, y2+, b2+ | N.D. | N.D. | N.D. | N.D. | |
| NGDSPITVPPNGYFR | 510–524 | 2 | 817.402 | 822.406 | y11+, y9+, y7+, b2+ | <0.19 | 1.53 | 50 | 19.38 | |
| GII.4_X76716/ Bristol | NNFYHYNQANDSTLK | 163–177 | 3 | 610.280 | 612.951 | y7+, y6+, y4+, b2+ | <0.19 | 6.1 | 6.25 | 23.05 |
| ANNAGDDVFTVSCR | 188–201 | 2 | 763.339 | 768.343 | y10+, y6+, y3+, b2+ | <0.19 | 6.1 | >100 | 21.92 | |
| GII.6_AJ277620/ Seacroft/90/UK | GTLISQTAR | 287–295 | 2 | 473.769 | 478.774 | y7+, y6+, y5+, y1+ | <0.19 | 1.53 | 6.25 | 15.95 |
| NHPLHVQVK | 307–315 | 3 | 357.873 | 360.545 | y5+, y4+, y1+, b2+ | N.D. | N.D. | N.D. | N.D. | |
| LGTILIK | 380–386 | 2 | 379.263 | 383.270 | y2+, y1+, b3+, b22+ | <0.19 | 3.05 | 0.39 | 19.53 | |
| GII.17_AY502009/ CS-E1/2002/US | NVGSNPNTTR | 340–349 | 2 | 530.263 | 535.267 | y8+, y5+, y1+, b2+ | N.D. | N.D. | N.D. | N.D. |
| FTPVGIK | 387–393 | 2 | 381.231 | 385.239 | y5+, y3+, y1+, b2+ | <0.19 | 6.1 | 12.5 | 19.65 |
| Patient No. | PCR Analysis | Extracted Total Protein (μg) | PRM Assay | ||
|---|---|---|---|---|---|
| Genotype | Ct Value | Detected Target Peptide | Concentration (fmol/μg) | ||
| 1 | GII.3 | 19 | 693 | GII.3 NGDSPITVPPNGYFR_2+ | 13.17 |
| GII.3 VFGVASQR_2+ | <LLOQ * | ||||
| 2 | GII.4 | 19 | 319 | GII.4 ANNAGDDVFTVSCR_2+ | <LLOQ * |
| 3 | GII.P16/GII.2 | 18 | 545 | GII.2 YAGALNLNTNLAPSVAPVFPGER_2+ | 5.99 |
| 4 | GII.P16/GII.2 | 19 | 101 | GII.2 YAGALNLNTNLAPSVAPVFPGER_2+ | <LLOQ * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimura, Y.; Shin, J.; Nakai, Y.; Takahashi, M.; Ino, Y.; Akiyama, T.; Goto, K.; Nagata, N.; Yamaoka, Y.; Miyakawa, K.; et al. Development of Parallel Reaction Monitoring Mass Spectrometry Assay for the Detection of Human Norovirus Major Capsid Protein. Viruses 2022, 14, 1416. https://doi.org/10.3390/v14071416
Kimura Y, Shin J, Nakai Y, Takahashi M, Ino Y, Akiyama T, Goto K, Nagata N, Yamaoka Y, Miyakawa K, et al. Development of Parallel Reaction Monitoring Mass Spectrometry Assay for the Detection of Human Norovirus Major Capsid Protein. Viruses. 2022; 14(7):1416. https://doi.org/10.3390/v14071416
Chicago/Turabian StyleKimura, Yayoi, Jihye Shin, Yusuke Nakai, Masaya Takahashi, Yoko Ino, Tomoko Akiyama, Keiko Goto, Noriko Nagata, Yutaro Yamaoka, Kei Miyakawa, and et al. 2022. "Development of Parallel Reaction Monitoring Mass Spectrometry Assay for the Detection of Human Norovirus Major Capsid Protein" Viruses 14, no. 7: 1416. https://doi.org/10.3390/v14071416
APA StyleKimura, Y., Shin, J., Nakai, Y., Takahashi, M., Ino, Y., Akiyama, T., Goto, K., Nagata, N., Yamaoka, Y., Miyakawa, K., Kimura, H., & Ryo, A. (2022). Development of Parallel Reaction Monitoring Mass Spectrometry Assay for the Detection of Human Norovirus Major Capsid Protein. Viruses, 14(7), 1416. https://doi.org/10.3390/v14071416