
Potential Autoimmunity Resulting from Molecular Mimicry between SARS-CoV-2 Spike and Human Proteins

 , , ,
, , , 
Abstract
:1. Introduction
2. Methods
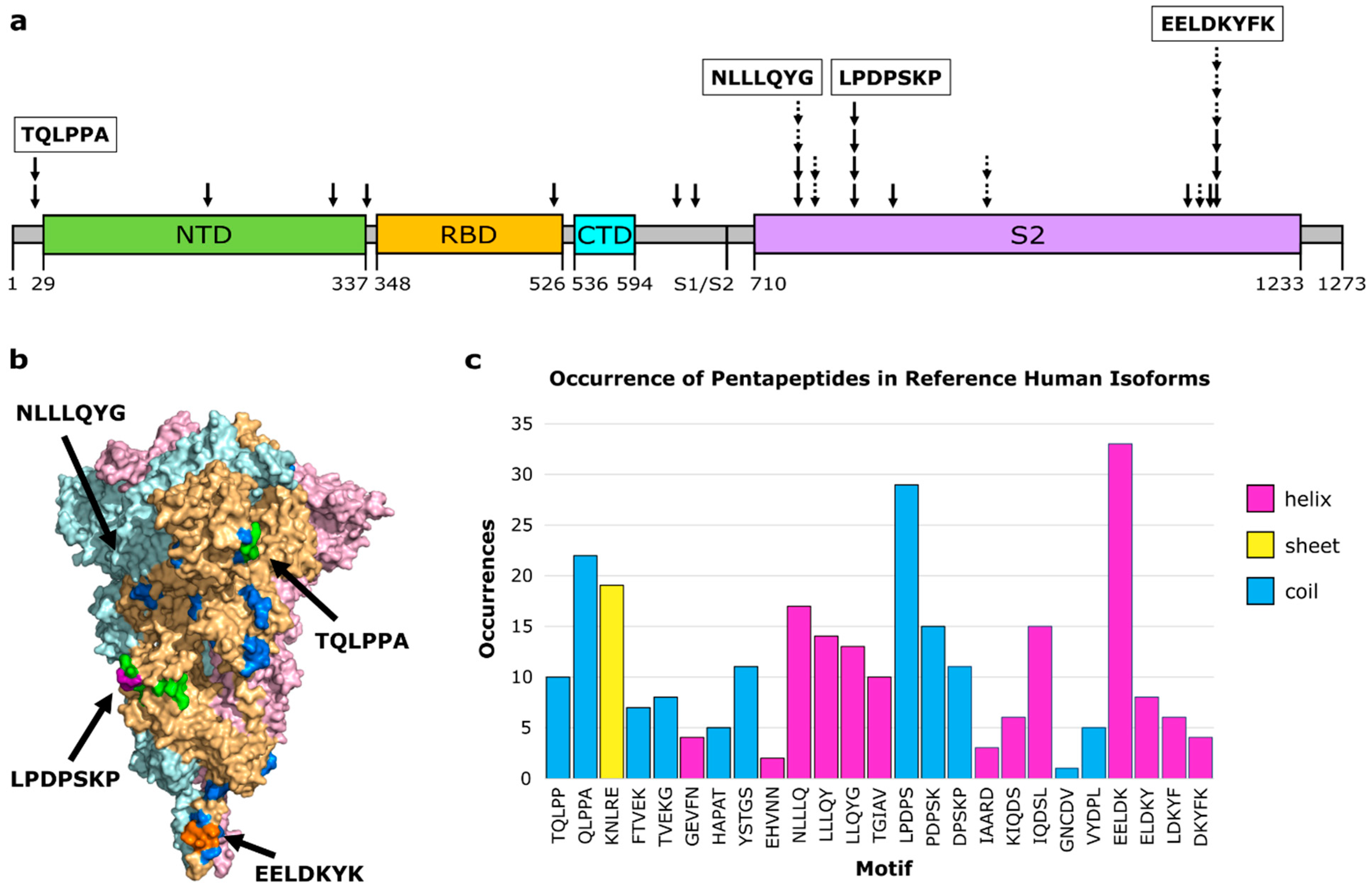
2.1. Identifying Epitopes with Molecular Mimicry
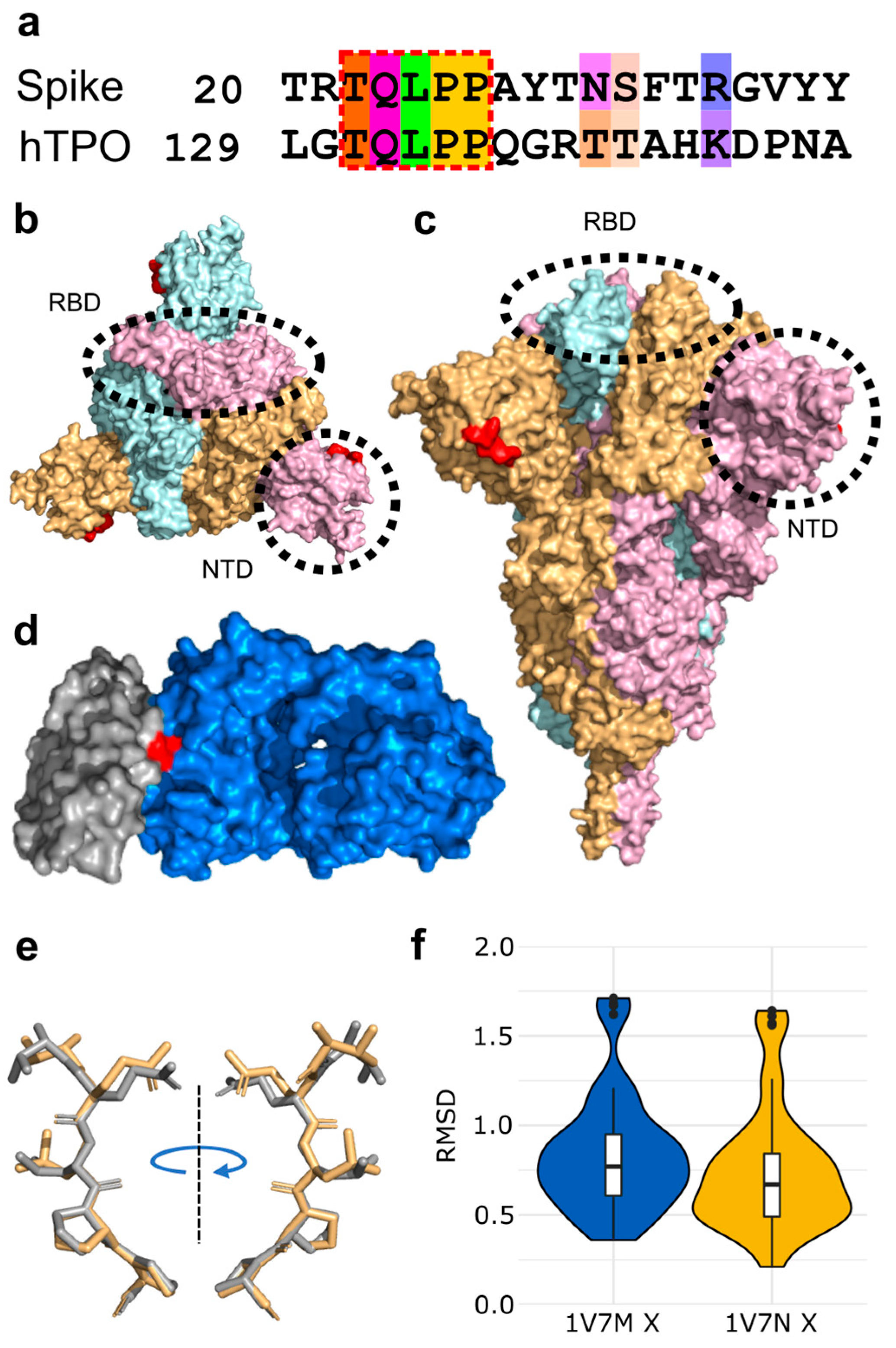
2.2. Conformational Ensemble of TQLPP Structural Mimicry
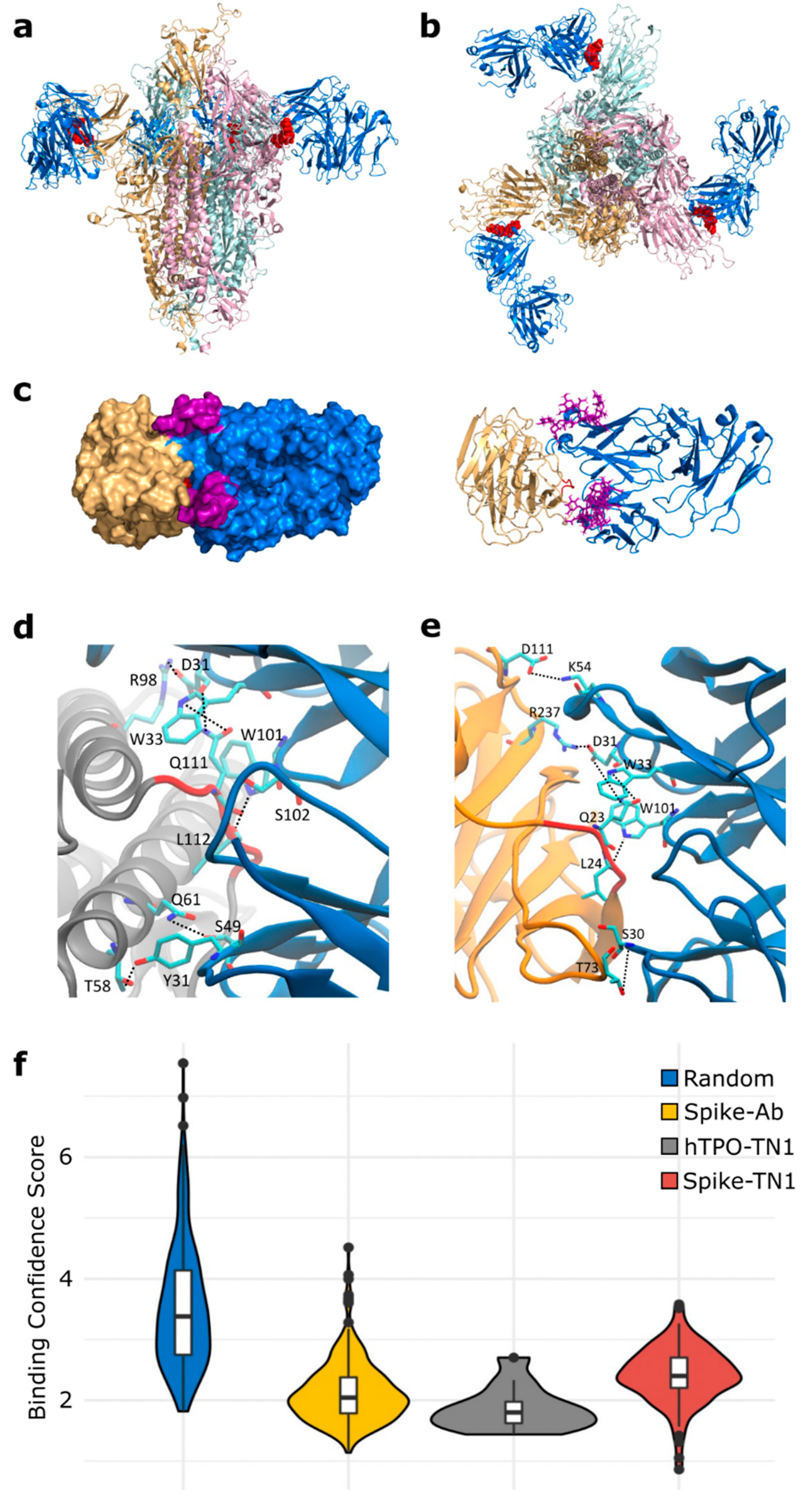
2.3. Modeling Spike-Antibody Complexes
2.4. Molecular Dynamics Simulation
2.5. Binding Affinity
2.6. Antibody Interface Complementarity
2.7. Evaluating Further Cross-Reactivity
2.8. Statistical Analysis
3. Results and Discussion
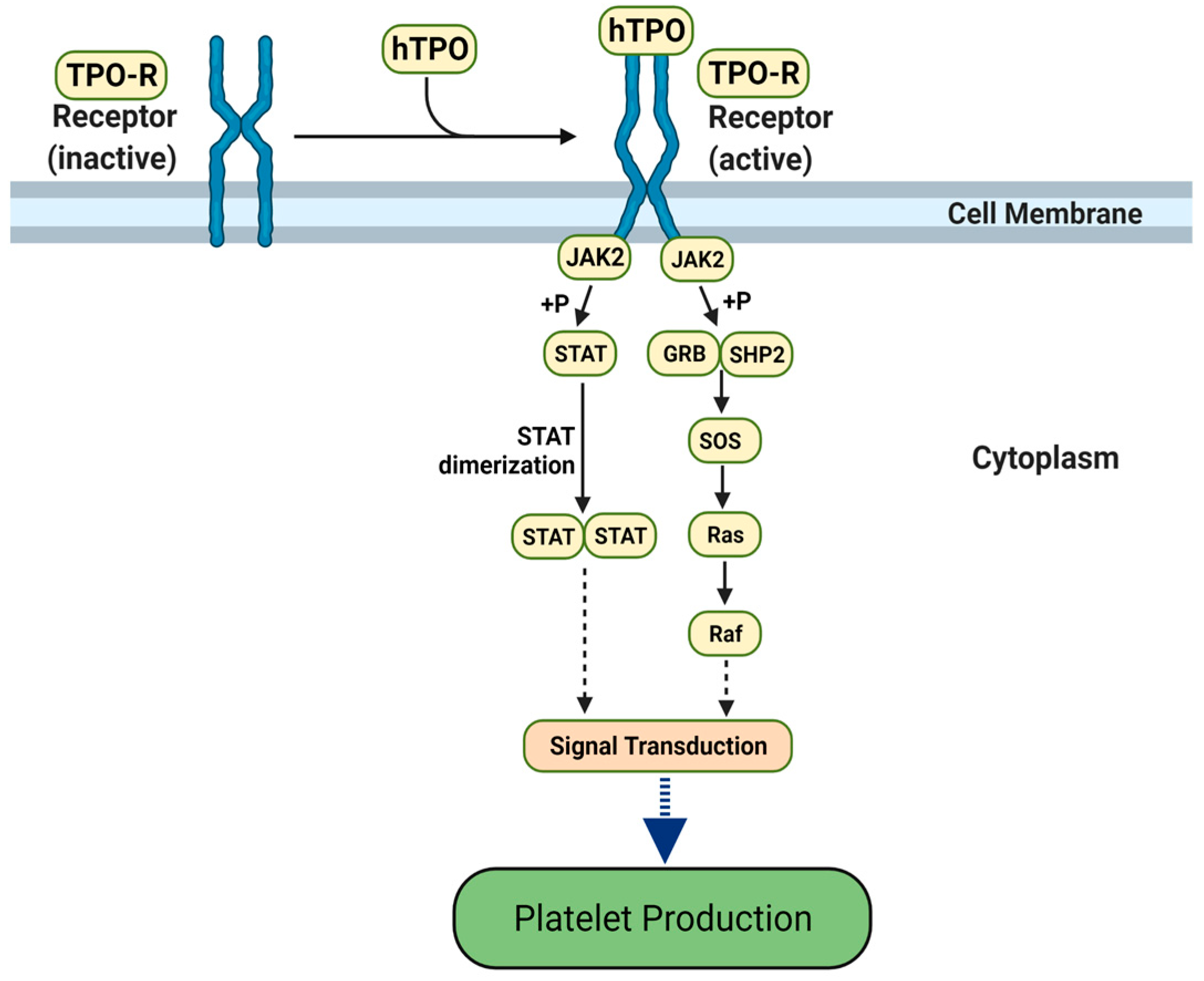
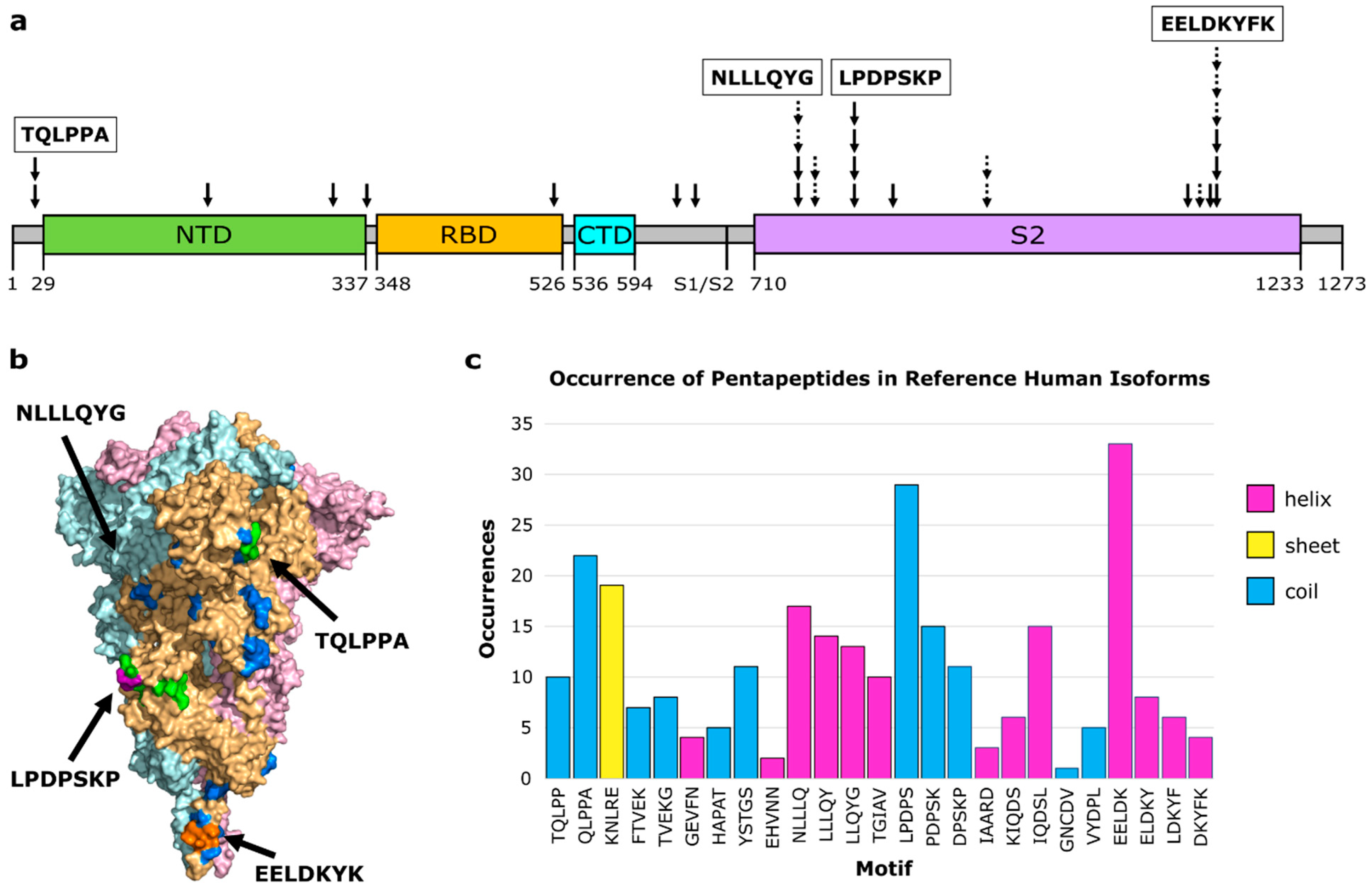
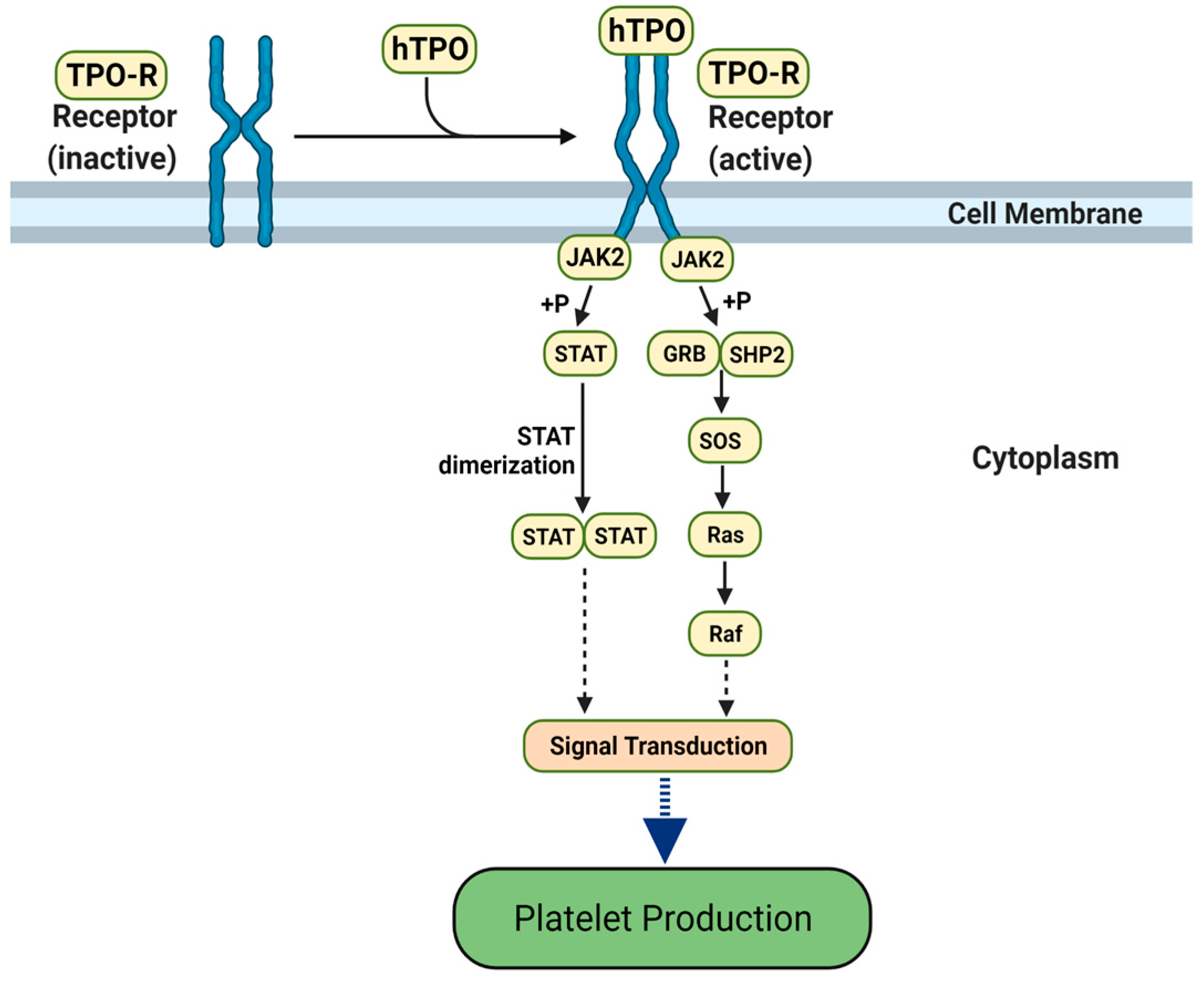
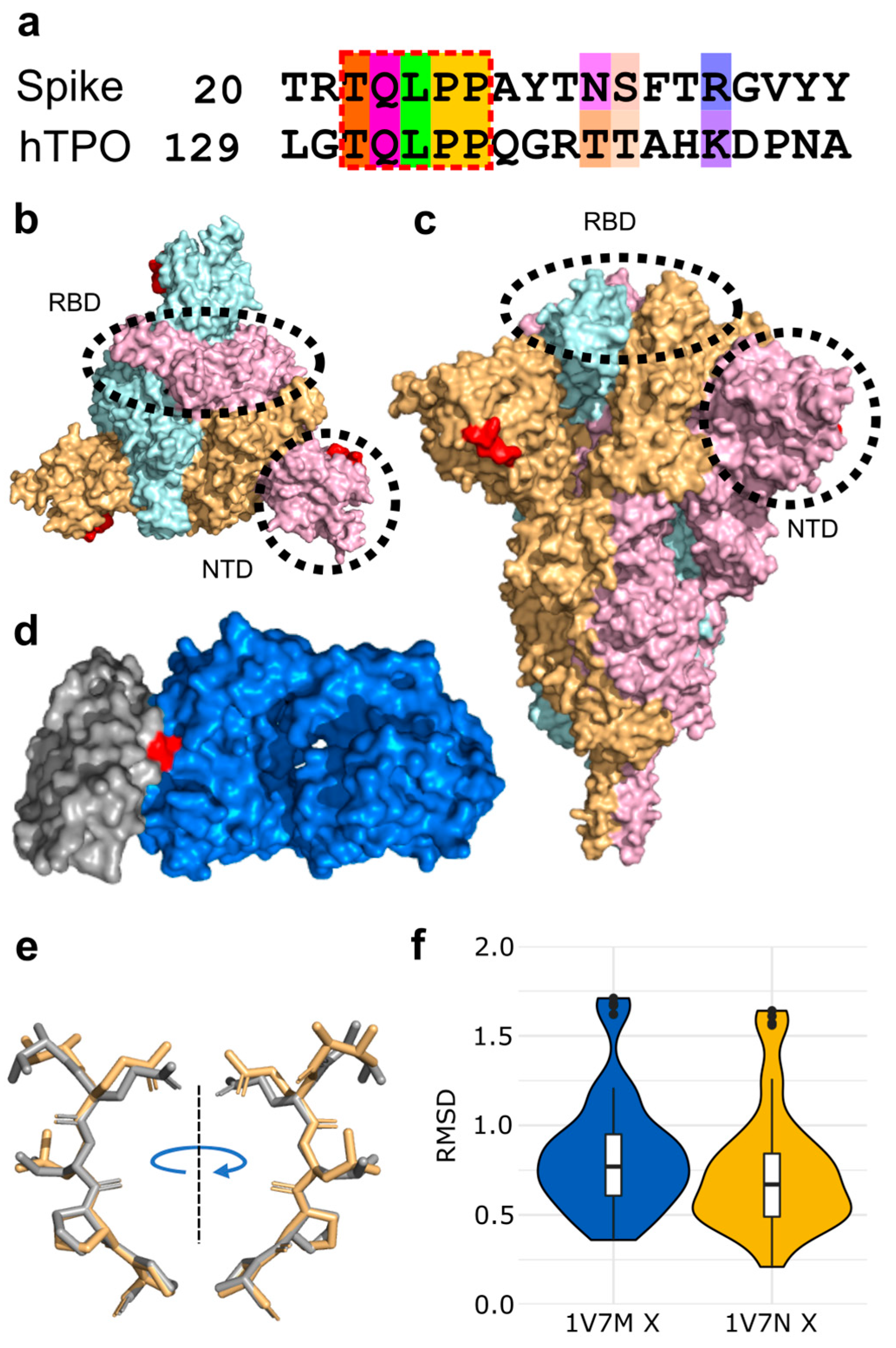
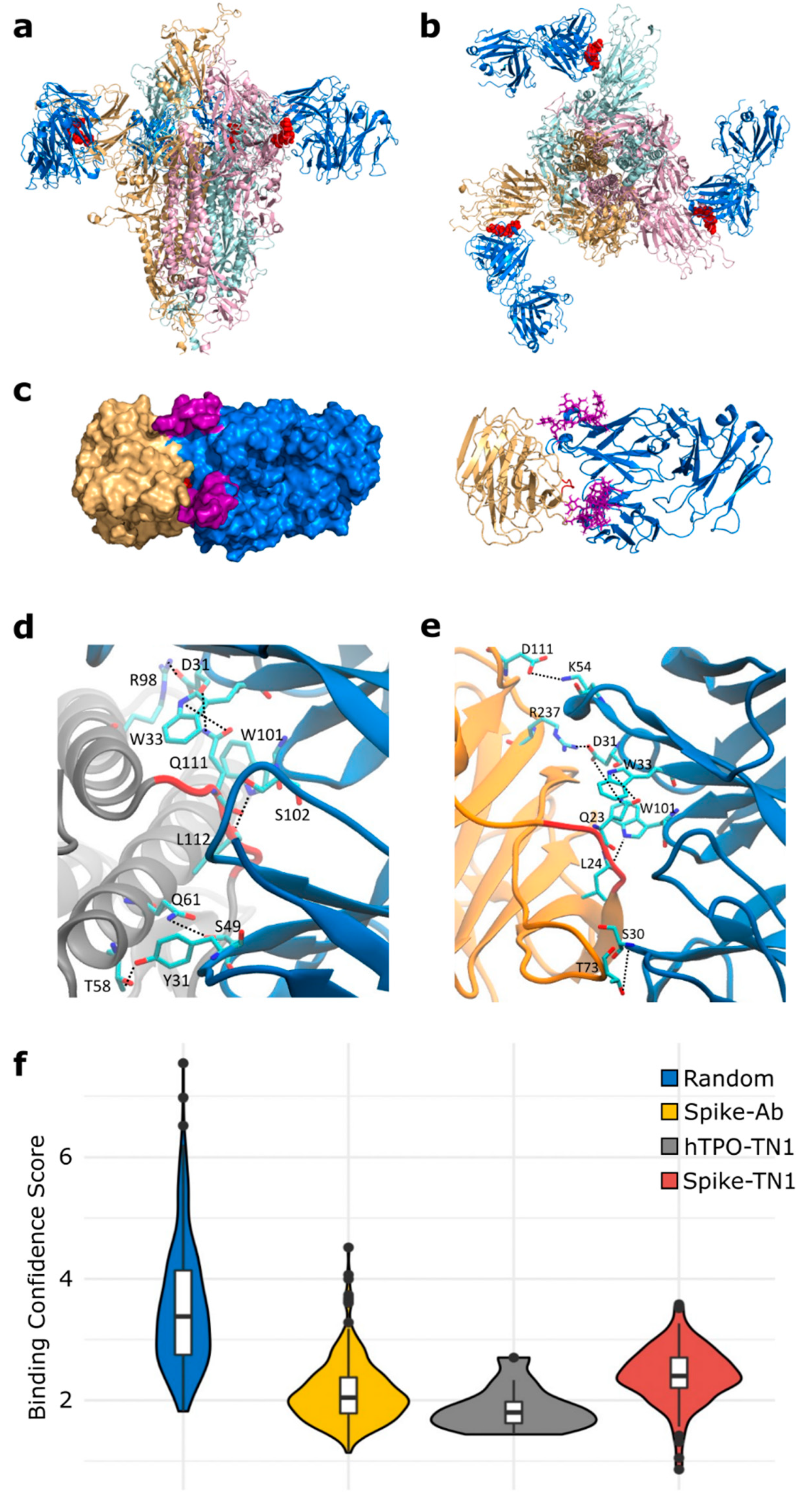
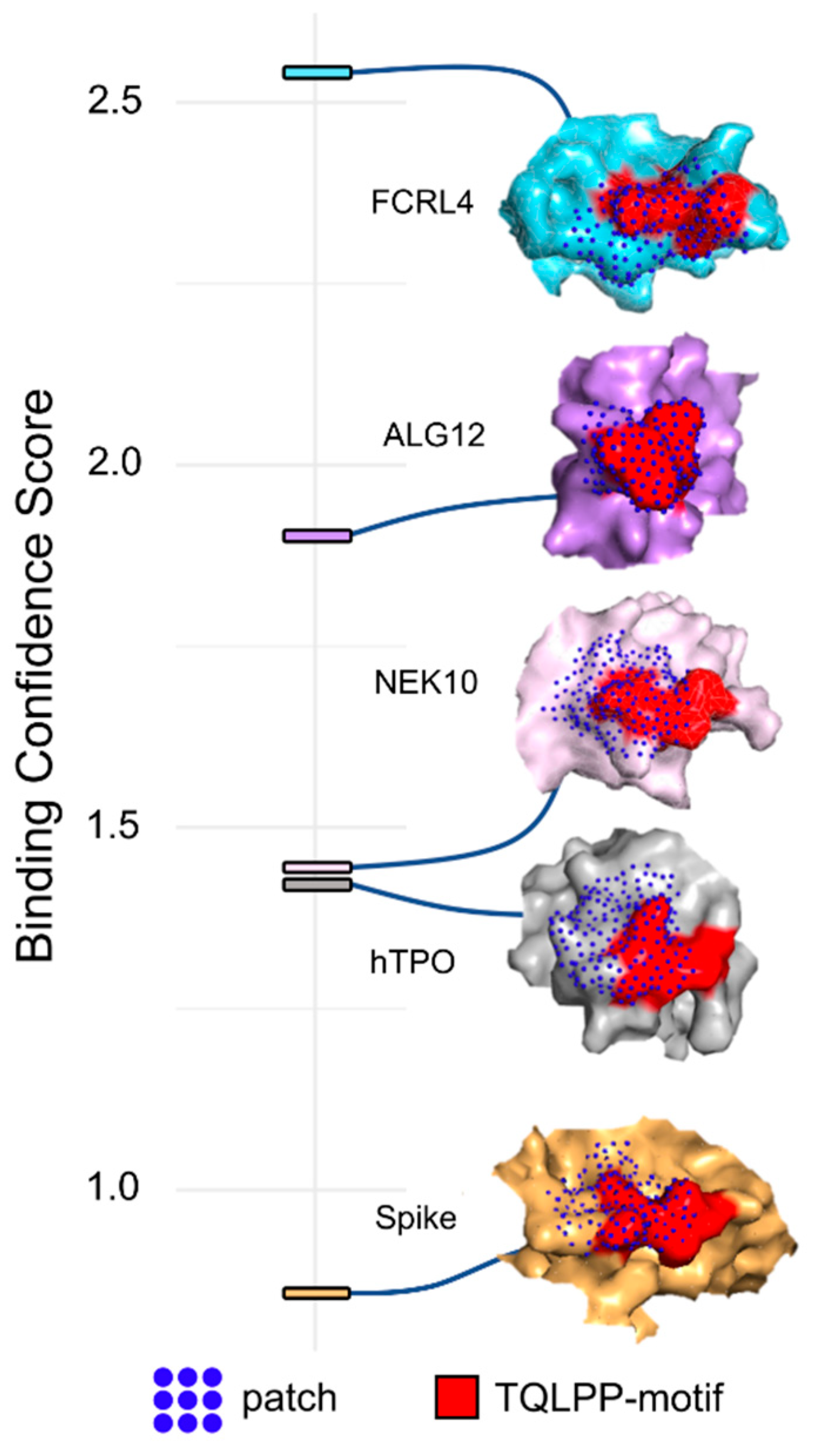
3.1. Molecular Mimicry between Spike and Thrombopoietin Mediated through TQLPP
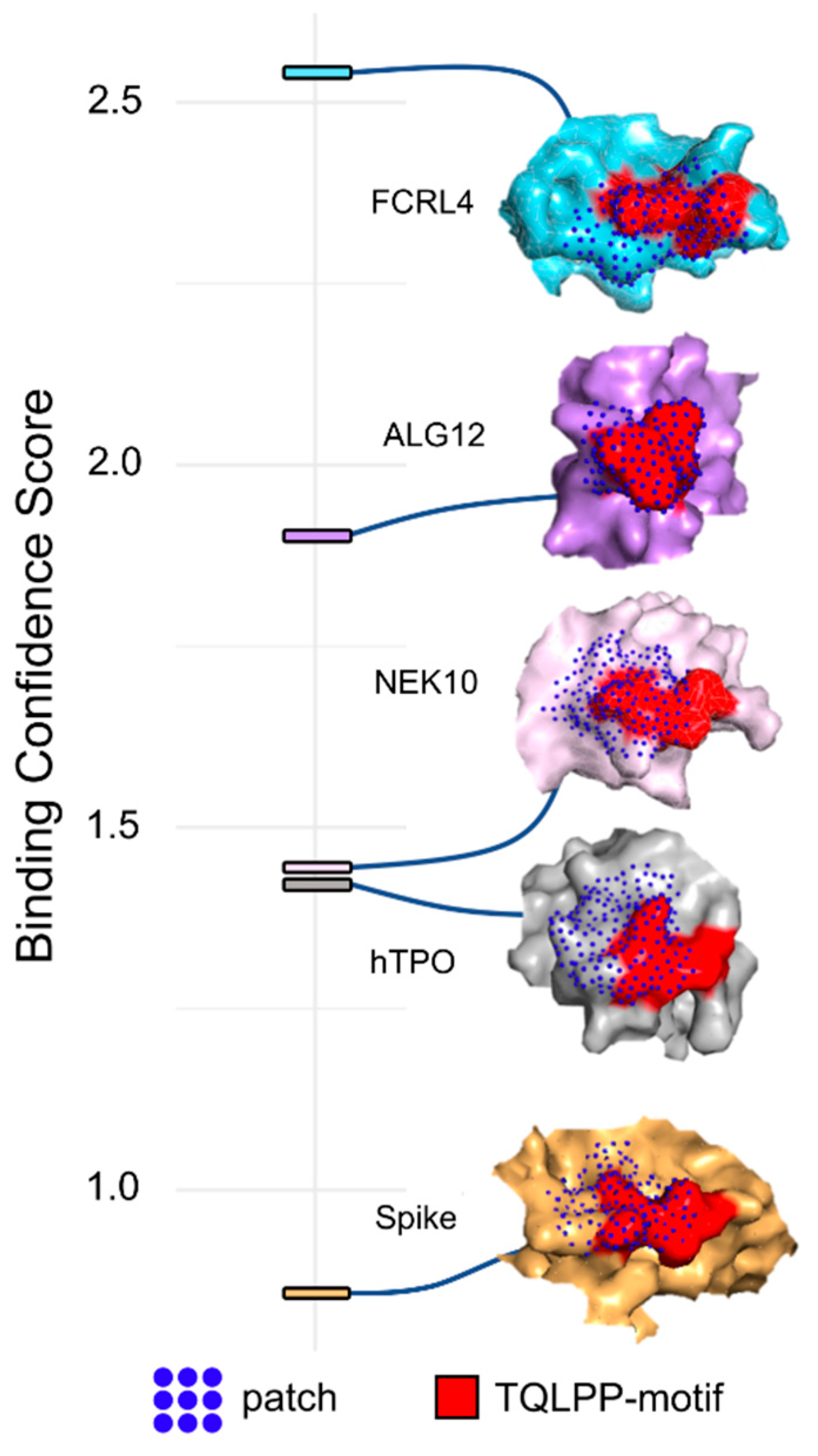
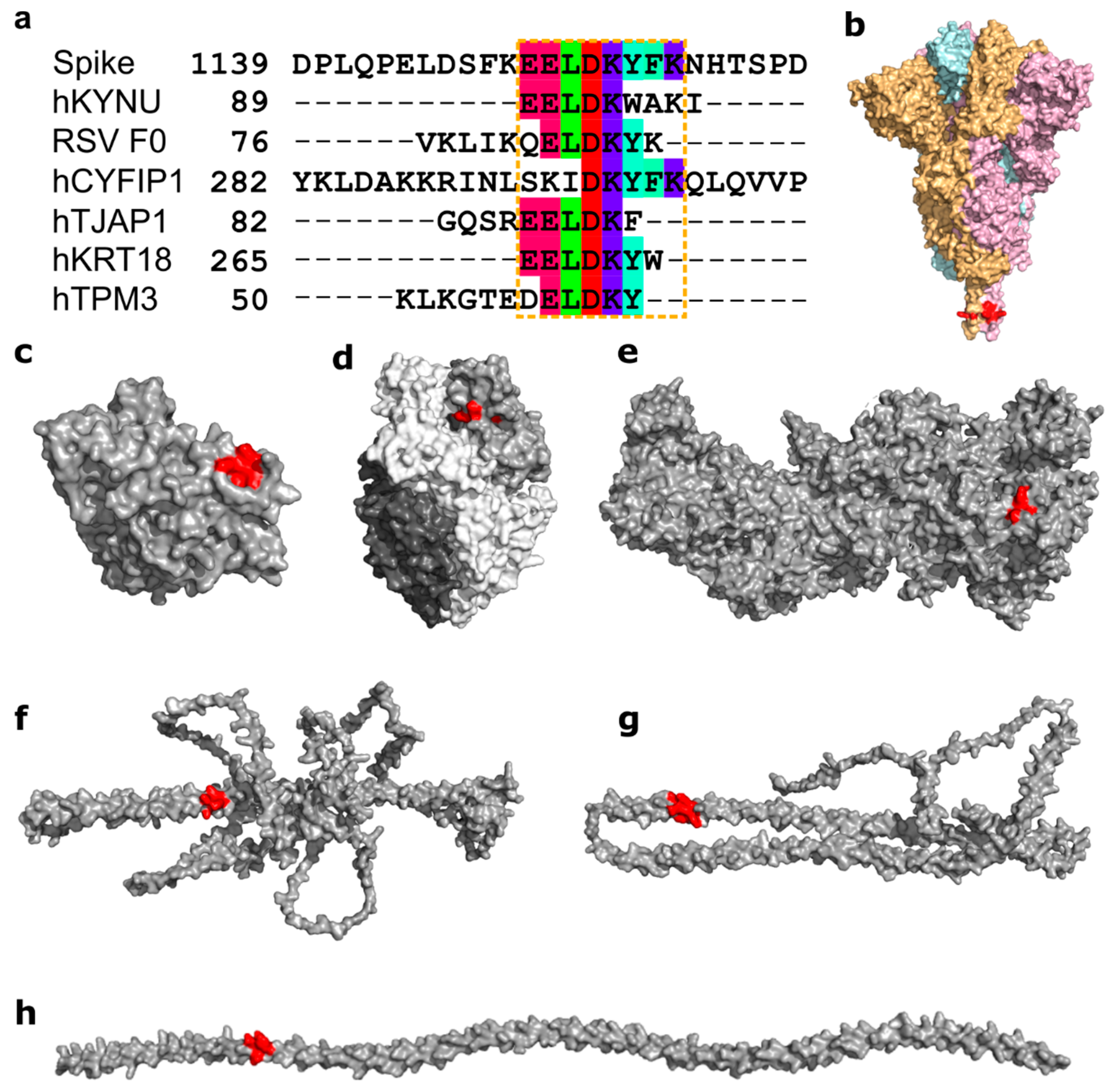
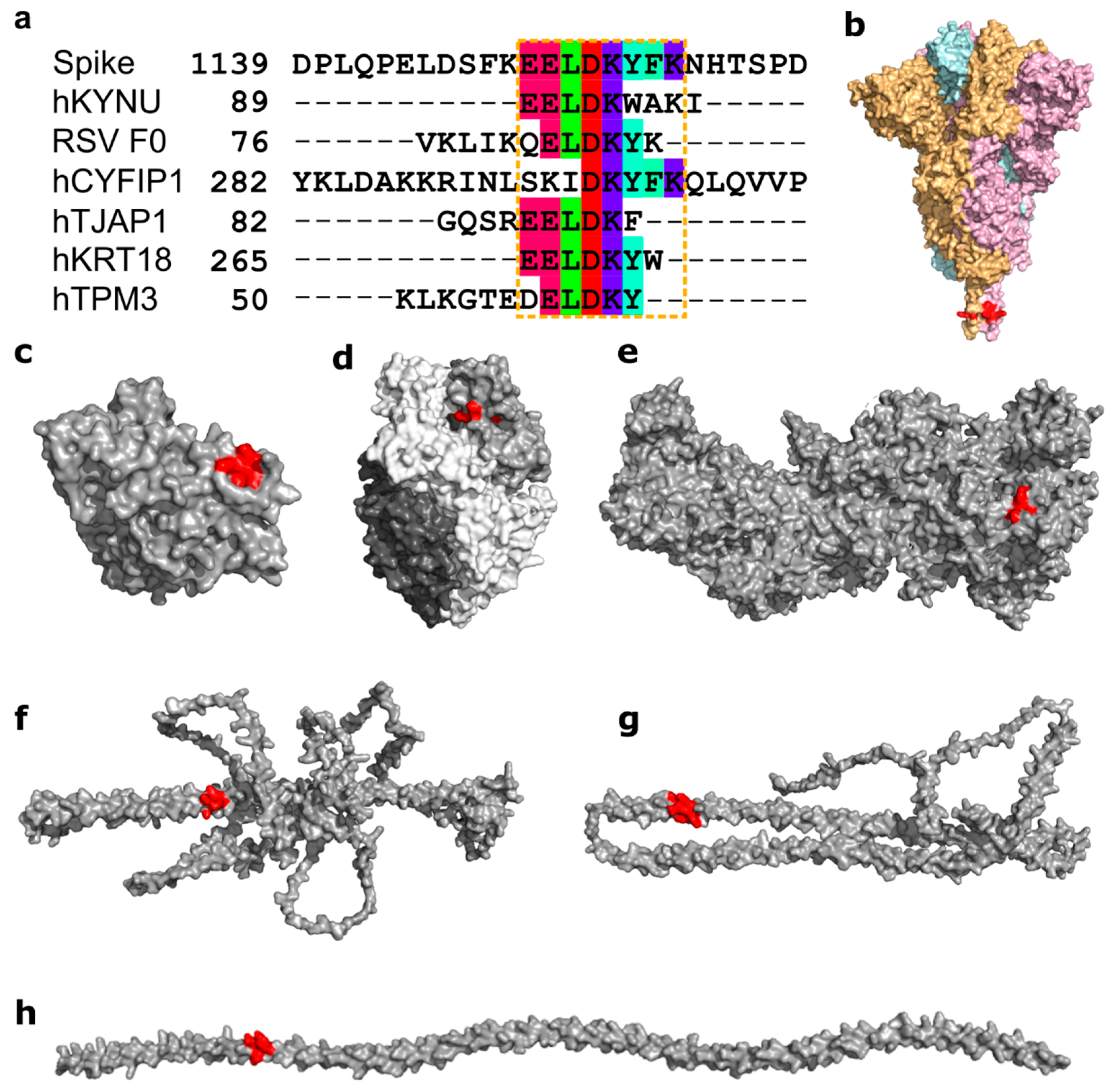
3.2. Molecular Mimicry between Spike, RSV, and Many Human Proteins Mediated through ELDKY
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.; Rabold, E.M.; Laws, R.L.; Conners, E.E.; Gharpure, R.; Yin, S.; Buono, S.A.; Dasu, T.; Bhattacharyya, S.; Westergaard, R.P.; et al. Loss of Taste and Smell as Distinguishing Symptoms of Coronavirus Disease. Clin. Infect. Dis. 2021, 72, 682–685. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An Interactive Web-Based Dashboard to Track COVID-19 in Real Time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Sah, P.; Fitzpatrick, M.C.; Zimmer, C.F.; Abdollahi, E.; Juden-Kelly, L.; Moghadas, S.M.; Singer, B.H.; Galvani, A.P. Asymptomatic SARS-CoV-2 Infection: A Systematic Review and Meta-Analysis. Proc. Natl. Acad. Sci. USA 2021, 118, e2109229118. [Google Scholar] [CrossRef] [PubMed]
- Saviano, A.; Wrensch, F.; Ghany, M.G.; Baumert, T.F. Liver Disease and Coronavirus Disease 2019: From Pathogenesis to Clinical Care. Hepatology 2021, 74, 1088–1100. [Google Scholar] [CrossRef]
- Han, X.; Ye, Q. Kidney Involvement in COVID-19 and Its Treatments. J. Med. Virol. 2021, 93, 1387–1395. [Google Scholar] [CrossRef]
- Long, B.; Brady, W.J.; Koyfman, A.; Gottlieb, M. Cardiovascular Complications in COVID-19. Am. J. Emerg. Med. 2020, 38, 1504–1507. [Google Scholar] [CrossRef]
- Mei, H.; Luo, L.; Hu, Y. Thrombocytopenia and Thrombosis in Hospitalized Patients with COVID-19. J. Hematol. Oncol. 2020, 13, 161. [Google Scholar] [CrossRef]
- Wei, J.; Matthews, P.C.; Stoesser, N.; Maddox, T.; Lorenzi, L.; Studley, R.; Bell, J.I.; Newton, J.N.; Farrar, J.; Diamond, I.; et al. Anti-Spike Antibody Response to Natural SARS-CoV-2 Infection in the General Population. Nat. Commun. 2021, 12, 6250. [Google Scholar] [CrossRef]
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; Wong, P.; et al. Diverse Functional Autoantibodies in Patients with COVID-19. Nature 2021, 595, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.R.; Chastain, E.M.; Terry, R.L.; Miller, S.D. Virus Infection, Antiviral Immunity, and Autoimmunity. Immunol. Rev. 2013, 255, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, B. Heterologous Immunity: Role in Natural and Vaccine-Induced Resistance to Infections. Front. Immunol. 2019, 10, 2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraley, E.; LeMaster, C.; Banerjee, D.; Khanal, S.; Selvarangan, R.; Bradley, T. Cross-Reactive Antibody Immunity against SARS-CoV-2 in Children and Adults. Cell. Mol. Immunol. 2021, 18, 1826–1828. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Voss, C.; Esmail, S.; Liu, X.; Knauer, M.J.; Ackloo, S.; Kaneko, T.; Lowes, L.; Stogios, P.; Seitova, A.; Hutchinson, A.; et al. Epitope-Specific Antibody Responses Differentiate COVID-19 Outcomes and Variants of Concern. JCI Insight 2021, 6, e148855. [Google Scholar] [CrossRef]
- Segal, Y.; Shoenfeld, Y. Vaccine-Induced Autoimmunity: The Role of Molecular Mimicry and Immune Crossreaction. Cell. Mol. Immunol. 2018, 15, 586–594. [Google Scholar] [CrossRef]
- Kanduc, D. From Anti-SARS-CoV-2 Immune Responses to COVID-19 via Molecular Mimicry. Antibodies 2020, 9, 33. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [Green Version]
- O’donoghue, S.I.; Schafferhans, A.; Sikta, N.; Stolte, C.; Kaur, S.; Ho, B.K.; Anderson, S.; Procter, J.B.; Dallago, C.; Bordin, N.; et al. SARS-CoV-2 Structural Coverage Map Reveals Viral Protein Assembly, Mimicry, and Hijacking Mechanisms. Mol. Syst. Biol. 2021, 17, e10079. [Google Scholar] [CrossRef]
- Khavinson, V.; Terekhov, A.; Kormilets, D.; Maryanovich, A. Homology between SARS-CoV-2 and Human Proteins. Sci. Rep. 2021, 11, 17199. [Google Scholar] [CrossRef] [PubMed]
- Balbin, C.A.; Nunez-Castilla, J.; Stebliankin, V.; Baral, P.; Sobhan, M.; Cickovski, T.; Mondal, A.M.; Narasimhan, G.; Chapagain, P.; Mathee, K.; et al. Epitopedia: Identifying Molecular Mimicry of Known Immune Epitopes. BioRxiv 2021. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct Conformational States of SARS-CoV-2 Spike Protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-Align: A Protein Structure Alignment Algorithm Based on the TM-Score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1255–1260. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K.; Cao, Y.; Frank, M.; Woo, H.; Park, S.J.; Yeom, M.S.; Croll, T.I.; Seok, C.; Im, W. Structure, Dynamics, Receptor Binding, and Antibody Binding of the Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein in a Viral Membrane. J. Chem. Theory Comput. 2021, 17, 2479–2487. [Google Scholar] [CrossRef]
- Pinto, D.; Sauer, M.M.; Czudnochowski, N.; Low, J.S.; Tortorici, M.A.; Housley, M.P.; Noack, J.; Walls, A.C.; Bowen, J.E.; Guarino, B.; et al. Broad Betacoronavirus Neutralization by a Stem Helix–Specific Human Antibody. Science 2021, 373, 1109–1116. [Google Scholar] [CrossRef]
- Schrödinger. The PyMOL Molecular Graphics System; Version 2.5.0; Schrödinger: New York, NY, USA, 2015. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosé, S.; Klein, M.L. Constant Pressure Molecular Dynamics for Molecular Systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A Web Server for Predicting the Binding Affinity of Protein-Protein Complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Gainza, P.; Sverrisson, F.; Monti, F.; Rodolà, E.; Boscaini, D.; Bronstein, M.M.; Correia, B.E. Deciphering Interaction Fingerprints from Protein Molecular Surfaces Using Geometric Deep Learning. Nat. Methods 2019, 17, 184–192. [Google Scholar] [CrossRef]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced Surface: An Efficient Way to Compute Molecular Surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
- Stebliankin, V.; Baral, P.; Balbin, C.; Nunez-Castilla, J.; Sobhan, M.; Cickovski, T.; Mohan Mondal, A.; Siltberg-Liberles, J.; Chapagain, P.; Mathee, K.; et al. EMoMiS: A Pipeline for Epitope-Based Molecular Mimicry Search in Protein Structures with Applications to SARS-CoV-2. BioRxiv 2022. [Google Scholar] [CrossRef]
- Dunbar, J.; Krawczyk, K.; Leem, J.; Baker, T.; Fuchs, A.; Georges, G.; Shi, J.; Deane, C.M. SAbDab: The Structural Antibody Database. Nucleic Acids Res. 2014, 42, D1140–D1146. [Google Scholar] [CrossRef] [PubMed]
- NCBI RefSeq Select. Available online: https://www.ncbi.nlm.nih.gov/refseq/refseq_select/ (accessed on 4 August 2021).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 173, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Premkumar, L.; Segovia-Chumbez, B.; Jadi, R.; Martinez, D.R.; Raut, R.; Markmann, A.J.; Cornaby, C.; Bartelt, L.; Weiss, S.; Park, Y.; et al. The Receptor-Binding Domain of the Viral Spike Protein Is an Immunodominant and Highly Specific Target of Antibodies in SARS-CoV-2 Patients. Sci. Immunol. 2020, 5, 8413. [Google Scholar] [CrossRef]
- Takeda, M. Proteolytic Activation of SARS-CoV-2 Spike Protein. Microbiol. Immunol. 2022, 66, 15–23. [Google Scholar] [CrossRef]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein Kinase CK2: A Potential Therapeutic Target for Diverse Human Diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712.e19. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The Protein Families Database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Varghese, L.N.; Defour, J.-P.; Pecquet, C.; Constantinescu, S.N. The Thrombopoietin Receptor: Structural Basis of Traffic and Activation by Ligand, Mutations, Agonists, and Mutated Calreticulin. Front. Endocrinol. 2017, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yang, Q.; Wang, Y.; Wu, Y.; Xu, J.; Yu, Y.; Shang, Y. Thrombocytopenia and Its Association with Mortality in Patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Wang, L.; Ye, J.; Gu, Z.; Wang, S.; Xia, J.; Xie, Y.; Li, Q.; Xu, R.; Lin, N. Predictors of Mortality in Patients with Coronavirus Disease 2019: A Systematic Review and Meta-Analysis. BMC Infect. Dis. 2021, 21, 663. [Google Scholar] [CrossRef] [PubMed]
- Nazy, I.; Kelton, J.G.; Moore, J.C.; Clare, R.; Horsewood, P.; Smith, J.W.; Ivetic, N.; D’Souza, V.; Li, N.; Arnold, D.M. Autoantibodies to Thrombopoietin and the Thrombopoietin Receptor in Patients with Immune Thrombocytopenia. Br. J. Haematol. 2018, 181, 234–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audia, S.; Bonnotte, B. Emerging Therapies in Immune Thrombocytopenia. J. Clin. Med. 2021, 10, 1004. [Google Scholar] [CrossRef] [PubMed]
- Watts, A.; Raj, K.; Gogia, P.; Gahona, C.C.T.; Porcelli, M. Secondary Immune Thrombocytopenic Purpura Triggered by COVID-19. Cureus 2021, 13, e14501. [Google Scholar] [CrossRef]
- Frankel, A.E.; Wylie, D.; Peters, B.; Marrama, D.; Ahn, C. Bioinformatic Analysis Underpinning the Frequent Occurence of Immune Thrombocytopenic Purpura in COVID-19 Patients. Isr. Med. Assoc. J. 2022, 24, 320–326. [Google Scholar]
- Mishra, N.; Huang, X.; Joshi, S.; Guo, C.; Ng, J.; Thakkar, R.; Wu, Y.; Dong, X.; Li, Q.; Pinapati, R.S.; et al. Immunoreactive Peptide Maps of SARS-CoV-2. Commun. Biol. 2021, 4, 225. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating Viruses and Cellular Organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Kuter, D.J. The Biology of Thrombopoietin and Thrombopoietin Receptor Agonists. Int. J. Hematol. 2013, 98, 10–23. [Google Scholar] [CrossRef]
- Feese, M.D.; Tamada, T.; Kato, Y.; Maeda, Y.; Hirose, M.; Matsukura, Y.; Shigematsu, H.; Muto, T.; Matsumoto, A.; Watarai, H.; et al. Structure of the Receptor-Binding Domain of Human Thrombopoietin Determined by Complexation with a Neutralizing Antibody Fragment. Proc. Natl. Acad. Sci. USA 2004, 101, 1816–1821. [Google Scholar] [CrossRef] [Green Version]
- Cerutti, G.; Guo, Y.; Zhou, T.; Gorman, J.; Lee, M.; Rapp, M.; Reddem, E.R.; Yu, J.; Bahna, F.; Bimela, J.; et al. Potent SARS-CoV-2 Neutralizing Antibodies Directed against Spike N-Terminal Domain Target a Single Supersite. Cell Host Microbe 2021, 29, 819–833.e7. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Kuwaki, T.; Matsumoto, A.; Morita, H.; Watarai, H.; Inagaki, Y.; Ohashi, H.; Ogami, K.; Miyazaki, H.; Kato, T. Neutralization of Biological Activity and Inhibition of Receptor Binding by Antibodies against Human Thrombopoietin. Stem Cells 1998, 16, 54–60. [Google Scholar] [CrossRef]
- Taylor, W.R. Residual Colours: A Proposal for Aminochromography. Protein Eng. 1997, 10, 743–746. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.; Park, S.J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I.; et al. Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [Google Scholar] [CrossRef] [PubMed]
- Kanduc, D. Thromboses and Hemostasis Disorders Associated with Coronavirus Disease 2019: The Possible Causal Role of Cross-Reactivity and Immunological Imprinting. Glob. Med. Genet. 2021, 8, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Chivukula, R.R.; Montoro, D.T.; Leung, H.M.; Yang, J.; Shamseldin, H.E.; Taylor, M.S.; Dougherty, G.W.; Zariwala, M.A.; Carson, J.; Daniels, L.A.; et al. A Human Ciliopathy Reveals Essential Functions for NEK10 in Airway Mucociliary Clearance. Nat. Med. 2020, 26, 244. [Google Scholar] [CrossRef]
- Andries, J.; Viranaicken, W.; Cordonin, C.; Herrscher, C.; Planesse, C.; Roquebert, B.; Lagrange-Xelot, M.; El-Kalamouni, C.; Meilhac, O.; Mavingui, P.; et al. The SARS-CoV-2 Spike Residues 616/644 and 1138/1169 Delineate Two Antibody Epitopes in COVID-19 MRNA COMINARTY Vaccine (Pfizer/BioNTech). Sci. Rep. 2022, 12, 5999. [Google Scholar] [CrossRef]
- Respiratory Syncytial Virus (RSV)|NIH: National Institute of Allergy and Infectious Diseases. Available online: https://www.niaid.nih.gov/diseases-conditions/respiratory-syncytial-virus-rsv (accessed on 8 January 2022).
- Li, Z.; Xi, X.; Gu, M.; Feil, R.; Ye, R.D.; Eigenthaler, M.; Hofmann, F.; Du, X. A Stimulatory Role for CGMP-Dependent Protein Kinase in Platelet Activation. Cell 2003, 112, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Sauzeau, V.; Le Jeune, H.; Cario-Toumaniantz, C.; Smolenski, A.; Lohmann, S.M.; Bertoglio, J.; Chardin, P.; Pacaud, P.; Loirand, G. Cyclic GMP-Dependent Protein Kinase Signaling Pathway Inhibits RhoA-Induced Ca2+ Sensitization of Contraction in Vascular Smooth Muscle. J. Biol. Chem. 2000, 275, 21722–21729. [Google Scholar] [CrossRef] [Green Version]
- Francis, S.H. The Role of CGMP-Dependent Protein Kinase in Controlling Cardiomyocyte CGMP. Circ. Res. 2010, 107, 1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, S.H.; Busch, J.L.; Corbin, J.D. CGMP-Dependent Protein Kinases and CGMP Phosphodiesterases in Nitric Oxide and CGMP Action. Pharmacol. Rev. 2010, 62, 525. [Google Scholar] [CrossRef] [PubMed]
- Szent-Györgyi, A.G. Calcium Regulation of Muscle Contraction. Biophys. J. 1975, 15, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Marrama, D.; Mahita, J.; Sette, A.; Peters, B. Lack of Evidence of Significant Homology of SARS-CoV-2 Spike Sequences to Myocarditis-Associated Antigens. EBioMedicine 2022, 75, 103807. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, H.; Liu, B.; Li, L.; Zhang, L.; Bao, M.; Ji, X.; He, X.; Yi, J.; Chen, P.; et al. Low Level Antibodies Against Alpha-Tropomyosin Are Associated With Increased Risk of Coronary Heart Disease. Front. Pharmacol. 2020, 11, 195. [Google Scholar] [CrossRef] [Green Version]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and Cardiovascular Disease: From Basic Mechanisms to Clinical Perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Patone, M.; Mei, X.W.; Handunnetthi, L.; Dixon, S.; Zaccardi, F.; Shankar-Hari, M.; Watkinson, P.; Khunti, K.; Harnden, A.; Coupland, C.A.C.; et al. Risks of Myocarditis, Pericarditis, and Cardiac Arrhythmias Associated with COVID-19 Vaccination or SARS-CoV-2 Infection. Nat. Med. 2021, 28, 410–422. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, E.; Bowe, B.; Al-Aly, Z. Long-Term Cardiovascular Outcomes of COVID-19. Nat. Med. 2022, 28, 583–590. [Google Scholar] [CrossRef]
- Li, Y.; Lai, D.; Zhang, H.; Jiang, H.; Tian, X.; Ma, M.; Qi, H.; Meng, Q.; Guo, S.; Wu, Y.; et al. Linear Epitopes of SARS-CoV-2 Spike Protein Elicit Neutralizing Antibodies in COVID-19 Patients. Cell. Mol. Immunol. 2020, 17, 1095–1097. [Google Scholar] [CrossRef]
- Stoddard, C.I.; Galloway, J.; Chu, H.Y.; Shipley, M.M.; Sung, K.; Itell, H.L.; Wolf, C.R.; Logue, J.K.; Magedson, A.; Garrett, M.E.; et al. Epitope Profiling Reveals Binding Signatures of SARS-CoV-2 Immune Response in Natural Infection and Cross-Reactivity with Endemic Human CoVs. Cell Rep. 2021, 35, 109164. [Google Scholar] [CrossRef]
- Helms, J.M.; Ansteatt, K.T.; Roberts, J.C.; Kamatam, S.; Foong, K.S.; Labayog, J.M.S.; Tarantino, M.D. Severe, Refractory Immune Thrombocytopenia Occurring after SARS-CoV-2 Vaccine. J. Blood Med. 2021, 12, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.H.; Sørvoll, I.H.; Michelsen, A.E.; Munthe, L.A.; Lund-Johansen, F.; Ahlen, M.T.; Wiedmann, M.; Aamodt, A.-H.; Skattør, T.H.; Tjønnfjord, G.E.; et al. Thrombosis and Thrombocytopenia after ChAdOx1 NCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic Thrombocytopenia after ChAdOx1 NCov-19 Vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B. CoVariants: SARS-CoV-2 Mutations and Variants of Interest. 2022. Available online: https://covariants.org/shared-mutations (accessed on 8 January 2022).
- Slabinski, L.; Jaroszewski, L.; Rodrigues, A.P.C.; Rychlewski, L.; Wilson, I.A.; Lesley, S.A.; Godzik, A. The Challenge of Protein Structure Determination—Lessons from Structural Genomics. Protein Sci. 2007, 16, 2472–2482. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Motif | Protein | Species | RMSD (Å) | Z-Score | EpiScore | PDB_Chain |
|---|---|---|---|---|---|---|
| TQLPP | Thrombopoietin | Human | 0.46 | −1.34 | 10.87 | 1V7N_X |
| QLPPA | SMYD3 protein | Human | 0.38 | −1.42 | 13.16 | 5CCL_A |
| KNLRE | Toll-like receptor 8 | Human | 0.87 | −0.92 | 5.75 | 6WML_D |
| FTVEKG | Pollen allergen Phl p2 | Phleum pratense | 0.76 | −1.03 | 7.89 | 1WHP_A |
| GEVFN | Integrin beta 1 | Human | 0.63 | −1.16 | 7.94 | 7NWL_B |
| HAPAT | Activator of 90 kDa heat shock protein ATPase homolog 1 | Human | 0.74 | −1.05 | 6.76 | 7DME_A |
| YSTGS | Argininosuccinate lyase | Human | 0.48 | −1.31 | 10.42 | 1K62_B |
| EHVNN | Casein kinase 2 alpha isoform | Human | 0.29 | −1.51 | 17.24 | 2ZJW_A |
| NLLLQ | DNA polymerase subunit gamma 1 | Human | 0.57 | −1.22 | 8.77 | 5C51_A |
| LLQYG | Ankyrin 1 | Human | 0.20 | −1.60 | 25.00 | 1N11_A |
| LPDPS | BRCA1-A complex subunit BRE | Human | 0.32 | −1.48 | 15.62 | 6GVW_C |
| LPDPS | Semaphorin 7a | Human | 0.84 | −0.91 | 5.95 | 3NVQ_A |
| DPSKP | 60S ribosomal protein L3 | Human | 0.10 | −1.70 | 50.00 | 6LU8_B |
| DPSKP | Alanine and proline-rich secreted protein apa precursor | Mycobacterium tuberculosis | 0.21 | −1.59 | 23.81 | 5ZXA_A |
| IAARD | Talin | Mus musculus | 0.74 | −1.05 | 6.76 | 6R9T_A |
| GNCDV | Tryptophan-tRNA ligase | Human | 0.91 | −0.88 | 5.49 | 1O5T_A |
| SFKEE | Small subunit processome component 20 homolog | Human | 0.32 | −1.48 | 15.62 | 7MQA_SP |
| EELDK | Kynureninase | Human | 0.22 | −1.58 | 22.73 | 2HZP_A |
| ELDKY | Fusion glycoprotein F0 | Respiratory syncytial virus | 0.12 | −1.68 | 41.67 | 6EAE_F |
| DKYFK | Cytoplasmic FMR1-interacting protein 1 | Human | 0.14 | −1.66 | 35.71 | 4N78_A |
| Motif | Protein | RMSD (Å) | Z-Score | EpiScore | AlphaFold2 ID |
|---|---|---|---|---|---|
| NLLLQ | Ankyrin 3 | 0.61 | −1.18 | 8.20 | AF-Q12955-F1-model_v1_A |
| LLQYG | Olfactory receptor 10Q1 | 0.66 | −1.13 | 7.58 | AF-Q8NGQ4-F1-model_v1_A |
| TGIAV | Phosphofructokinase | 0.17 | −1.63 | 29.41 | AF-P17858-F1-model_v1_A |
| TGIAV | Low affinity immunoglobulin gamma Fc region receptor II-b | 0.17 | −1.63 | 29.41 | AF-P31995-F1-model_v1_A |
| KIQDSL | Phosphorylase b kinase regulatory subunit beta | 0.19 | −1.61 | 31.58 | AF-Q93100-F1-model_v1_A |
| KIQDSL | Long-chain-fatty-acid-CoA ligase 5 | 0.37 | −1.43 | 16.22 | AF-Q9ULC5-F1-model_v1_A |
| VYDPL | Actin-binding protein IPP | 0.17 | −1.63 | 29.41 | AF-Q9Y573-F1-model_v1_A |
| EELDK | Tight junction-associated protein 1 | 0.20 | −1.60 | 25.00 | AF-Q5JTD0-F1-model_v1_A |
| EELDKY | Keratin, type I cytoskeletal 18 | 0.22 | −1.58 | 27.27 | AF-P05783-F1-model_v1_A |
| ELDKY | Tropomyosin alpha-3 chain | 0.18 | −1.62 | 27.78 | AF-P06753-F1-model_v1_A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunez-Castilla, J.; Stebliankin, V.; Baral, P.; Balbin, C.A.; Sobhan, M.; Cickovski, T.; Mondal, A.M.; Narasimhan, G.; Chapagain, P.; Mathee, K.; et al. Potential Autoimmunity Resulting from Molecular Mimicry between SARS-CoV-2 Spike and Human Proteins. Viruses 2022, 14, 1415. https://doi.org/10.3390/v14071415
Nunez-Castilla J, Stebliankin V, Baral P, Balbin CA, Sobhan M, Cickovski T, Mondal AM, Narasimhan G, Chapagain P, Mathee K, et al. Potential Autoimmunity Resulting from Molecular Mimicry between SARS-CoV-2 Spike and Human Proteins. Viruses. 2022; 14(7):1415. https://doi.org/10.3390/v14071415
Chicago/Turabian StyleNunez-Castilla, Janelle, Vitalii Stebliankin, Prabin Baral, Christian A. Balbin, Masrur Sobhan, Trevor Cickovski, Ananda Mohan Mondal, Giri Narasimhan, Prem Chapagain, Kalai Mathee, and et al. 2022. "Potential Autoimmunity Resulting from Molecular Mimicry between SARS-CoV-2 Spike and Human Proteins" Viruses 14, no. 7: 1415. https://doi.org/10.3390/v14071415
APA StyleNunez-Castilla, J., Stebliankin, V., Baral, P., Balbin, C. A., Sobhan, M., Cickovski, T., Mondal, A. M., Narasimhan, G., Chapagain, P., Mathee, K., & Siltberg-Liberles, J. (2022). Potential Autoimmunity Resulting from Molecular Mimicry between SARS-CoV-2 Spike and Human Proteins. Viruses, 14(7), 1415. https://doi.org/10.3390/v14071415