
Detection of Ancient Viruses and Long-Term Viral Evolution


Abstract
:1. Introduction
2. History of Ancient Viral Studies
3. Samples for Ancient Virus Discoveries and How to Analyze Them
3.1. Historical Samples
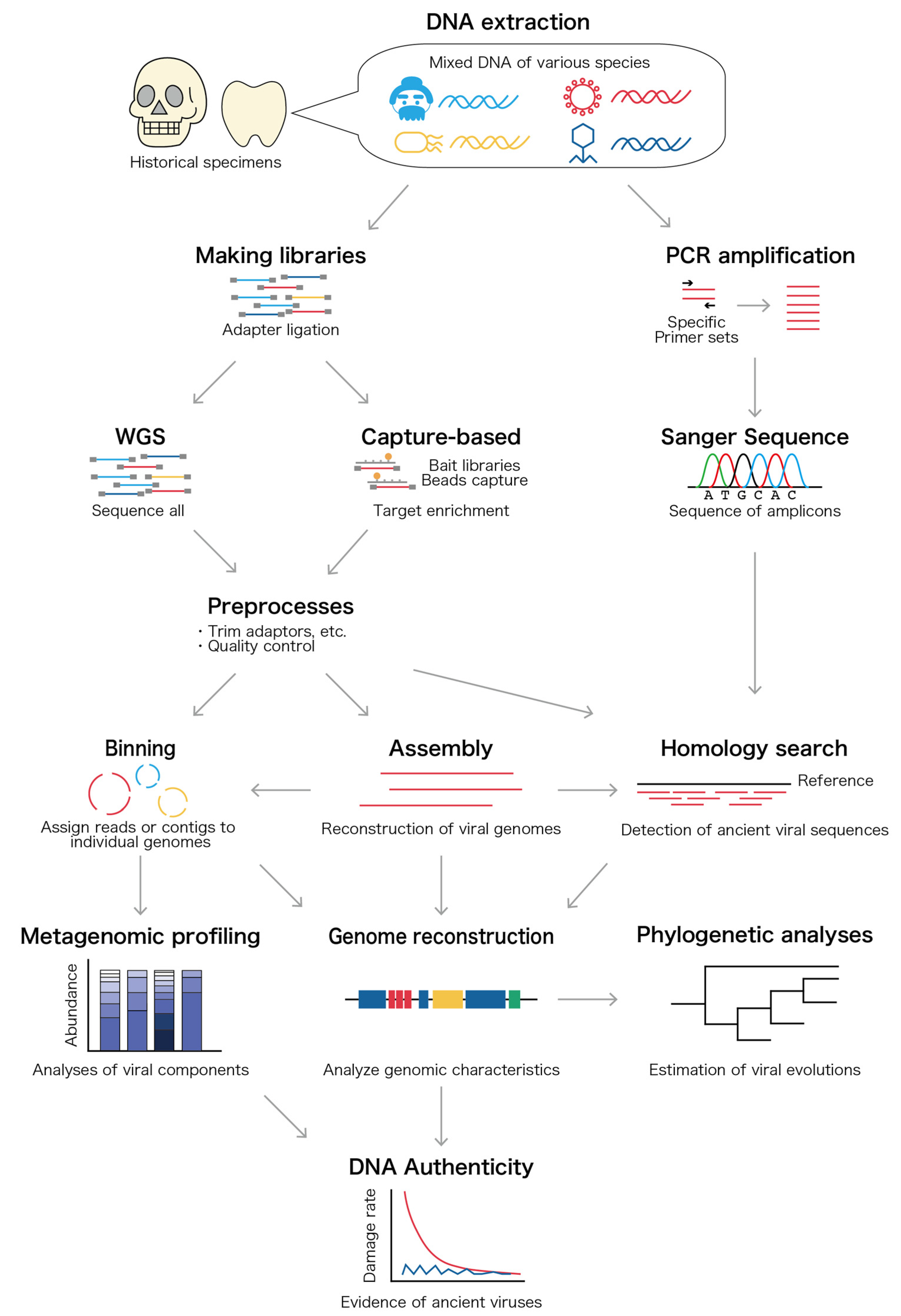
3.2. DNA and RNA Extraction, Amplification, and Sequencing
3.3. Bioinformatic Analysis
4. Metagenomic Data to Comprehend the Ancient Human Virome
5. Specific Ancient Viral Studies Inferring Past Pandemic and Evolutionary History
6. Long-Term Viral Evolution Reflecting Time-Dependent Rate Phenomenon (TDRP) Elucidated by Ancient Viral Sequences
7. Future Perspective: Detecting Highly Diverged or Extinctic Viral Genomes
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higuchi, R.; Bowman, B.; Freiberger, M.; Ryder, O.A.; Wilson, A.C. DNA Sequences from the Quagga, an Extinct Member of the Horse Family. Nature 1984, 312, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Prüfer, K.; Racimo, F.; Patterson, N.; Jay, F.; Sankararaman, S.; Sawyer, S.; Heinze, A.; Renaud, G.; Sudmant, P.H.; De Filippo, C.; et al. The Complete Genome Sequence of a Neanderthal from the Altai Mountains. Nature 2014, 505, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.; Renaud, G.; Viola, B.; Hublin, J.J.; Gansauge, M.T.; Shunkov, M.V.; Derevianko, A.P.; Prüfer, K.; Kelso, J.; Pääbo, S. Nuclear and Mitochondrial DNA Sequences from Two Denisovan Individuals. Proc. Natl. Acad. Sci. USA 2015, 112, 15696–15700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.; Kircher, M.; Gansauge, M.T.; Li, H.; Racimo, F.; Mallick, S.; Schraiber, J.G.; Jay, F.; Prüfer, K.; De Filippo, C.; et al. A High-Coverage Genome Sequence from an Archaic Denisovan Individual. Science 2012, 338, 222–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, S.R.; Browning, B.L.; Zhou, Y.; Tucci, S.; Akey, J.M. Analysis of Human Sequence Data Reveals Two Pulses of Archaic Denisovan Admixture. Cell 2018, 173, 53–61.e9. [Google Scholar] [CrossRef] [Green Version]
- Slon, V.; Mafessoni, F.; Vernot, B.; de Filippo, C.; Grote, S.; Viola, B.; Hajdinjak, M.; Peyrégne, S.; Nagel, S.; Brown, S.; et al. The Genome of the Offspring of a Neanderthal Mother and a Denisovan Father. Nature 2018, 561, 113–116. [Google Scholar] [CrossRef]
- Kanzawa-Kiriyama, H.; Jinam, T.A.; Kawai, Y.; Sato, T.; Hosomichi, K.; Tajima, A.; Adachi, N.; Matsumura, H.; Kryukov, K.; Saitou, N.; et al. Late Jomon Male and Female Genome Sequences from the Funadomari Site in Hokkaido, Japan. Anthropol. Sci. 2019, 127, 83–108. [Google Scholar] [CrossRef] [Green Version]
- Warinner, C.; Rodrigues, J.F.M.; Vyas, R.; Trachsel, C.; Shved, N.; Grossmann, J.; Radini, A.; Hancock, Y.; Tito, R.Y.; Fiddyment, S.; et al. Pathogens and Host Immunity in the Ancient Human Oral Cavity. Nat. Genet. 2014, 46, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Appelt, S.; Fancello, L.; Le Bailly, M.; Raoult, D.; Drancourt, M.; Desnues, C. Viruses in a 14th-Century Coprolite. Appl. Environ. Microbiol. 2014, 80, 2648–2655. [Google Scholar] [CrossRef] [Green Version]
- Warinner, C.; Speller, C.; Collins, M.J. A New Era in Palaeomicrobiology: Prospects for Ancient Dental Calculus as a Long-Term Record of the Human Oral Microbiome. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20130376. [Google Scholar] [CrossRef] [Green Version]
- Weyrich, L.S.; Duchene, S.; Soubrier, J.; Arriola, L.; Llamas, B.; Breen, J.; Morris, A.G.; Alt, K.W.; Caramelli, D.; Dresely, V.; et al. Neanderthal Behaviour, Diet, and Disease Inferred from Ancient DNA in Dental Calculus. Nature 2017, 544, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Reid, A.H.; Krafft, A.E.; Bijwaard, K.E.; Fanning, T.G. Initial Genetic Characterization of the 1918 “Spanish” Influenza Virus. Science 1997, 275, 1793–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, K.I.; Schuenemann, V.J.; Golding, G.B.; Burbano, H.A.; Waglechner, N.; Coombes, B.K.; McPhee, J.B.; Dewitte, S.N.; Meyer, M.; Schmedes, S.; et al. A Draft Genome of Yersinia Pestis from Victims of the Black Death. Nature 2011, 478, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Mühlemann, B.; Jones, T.C.; De Barros Damgaard, P.; Allentoft, M.E.; Shevnina, I.; Logvin, A.; Usmanova, E.; Panyushkina, I.P.; Boldgiv, B.; Bazartseren, T.; et al. Ancient Hepatitis B Viruses from the Bronze Age to the Medieval Period. Nature 2018, 557, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Krause-Kyora, B.; Susat, J.; Key, F.M.; Kühnert, D.; Bosse, E.; Immel, A.; Rinne, C.; Kornell, S.C.; Yepes, D.; Franzenburg, S.; et al. Neolithic and Medieval Virus Genomes Reveal Complex Evolution of Hepatitis B. eLife 2018, 7, e36666. [Google Scholar] [CrossRef]
- Spyrou, M.A.; Bos, K.I.; Herbig, A.; Krause, J. Ancient Pathogen Genomics as an Emerging Tool for Infectious Disease Research. Nat. Rev. Genet. 2019, 20, 323–340. [Google Scholar] [CrossRef]
- Duchêne, S.; Ho, S.Y.W.; Carmichael, A.G.; Holmes, E.C.; Poinar, H. The Recovery, Interpretation and Use of Ancient Pathogen Genomes. Curr. Biol. 2020, 30, R1215–R1231. [Google Scholar] [CrossRef]
- Simmonds, P.; Aiewsakun, P.; Katzourakis, A. Prisoners of War—Host Adaptation and Its Constraints on Virus Evolution. Nat. Rev. Microbiol. 2019, 17, 321–328. [Google Scholar] [CrossRef]
- Harvey, E.; Holmes, E.C. Diversity and Evolution of the Animal Virome. Nat. Rev. Microbiol. 2022, 1993, 30–33. [Google Scholar] [CrossRef]
- Virgin, H.W. The Virome in Mammalian Physiology and Disease. Cell 2014, 157, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The Human Gut Virome: Inter-Individual Variation and Dynamic Response to Diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadwell, K. The Virome in Host Health and Disease. Immunity 2015, 42, 805–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsangaras, K.; Greenwood, A.D. Paleovirology: Viral Sequences from Historical and Ancient DNA. In Paleogenomics: Genome-Scale Analysis of Ancient DNA; Lindqvist, C., Rajora, O.P., Eds.; Springer International Publishing: Cham, The Netherlands, 2018; pp. 139–162. ISBN 978-3-030-04753-5. [Google Scholar]
- Emerman, M.; Malik, H.S. Paleovirology—Modern Consequences of Ancient Viruses. PLoS Biol. 2010, 8, e1000301. [Google Scholar] [CrossRef] [PubMed]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific Enzymatic Amplification of DNA In Vitro: The Polymerase Chain Reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Korber, B.T.; Nahmias, A.J.; Hooper, E.; Sharp, P.M.; Ho, D.D. An African HIV-1 Sequence from 1959 and Implications for the of the Epidemic. Nature 1998, 391, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of Age: Ten Years of next-Generation Sequencing Technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Poinar, H.N.; Schwarz, C.; Qi, J.; Shapiro, B.; MacPhee, R.D.E.; Buigues, B.; Tikhonov, A.; Huson, D.H.; Tomsho, L.P.; Auch, A.; et al. Metagenomics to Paleogenomics: Large-Scale Sequencing of Mammoth DNA. Science 2006, 311, 392–394. [Google Scholar] [CrossRef] [Green Version]
- Smith, O.; Clapham, A.; Rose, P.; Liu, Y.; Wang, J.; Allaby, R.G. A Complete Ancient RNA Genome: Identification, Reconstruction and Evolutionary History of Archaeological Barley Stripe Mosaic Virus. Sci. Rep. 2014, 4, 4003. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, L.; Sugimoto, R.; Inoue, J.; Nakaoka, H.; Kanzawa-Kiriyama, H.; Shinoda, K.; Inoue, I. Identification of Ancient Viruses from Metagenomic Data of the Jomon People. J. Hum. Genet. 2021, 66, 287–296. [Google Scholar] [CrossRef]
- Hagelberg, E.; Sykes, B.; Hedges, R. Ancient Bone DNA Amplified. Nature 1989, 342, 485. [Google Scholar] [CrossRef]
- Seguin-Orlando, A.; Gamba, C.; der Sarkissian, C.; Ermini, L.; Louvel, G.; Boulygina, E.; Sokolov, A.; Nedoluzhko, A.; Lorenzen, E.D.; Lopez, P.; et al. Pros and Cons of Methylation-Based Enrichment Methods for Ancient DNA. Sci. Rep. 2015, 5, 11826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahila Bar-Gal, G.; Kim, M.J.; Klein, A.; Shin, D.H.; Oh, C.S.; Kim, J.W.; Kim, T.H.; Kim, S.B.; Grant, P.R.; Pappo, O.; et al. Tracing Hepatitis B Virus to the 16th Century in a Korean Mummy. Hepatology 2012, 56, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Peyambari, M.; Warner, S.; Stoler, N.; Rainer, D.; Roossinck, M.J. A 1000-Year-Old RNA Virus. J. Virol. 2019, 93, e01188-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, B.A.; Jones, R.A.C. Genetic Variability in the Coat Protein Gene of Potato Virus X and the Current Relationship between Phylogenetic Placement and Resistance Groupings. Arch. Virol. 2010, 155, 1349–1356. [Google Scholar] [CrossRef]
- Dullemans, A.M.; Cuperus, C.; Verbeek, M.; van der Vlugt, R.A.A. Complete Nucleotide Sequence of a Potato Isolate of Strain Group C of Potato Virus Y from 1938. Arch. Virol. 2011, 156, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Kehoe, M.A.; Jones, R.A.C. Improving Potato Virus Y Strain Nomenclature: Lessons from Comparing Isolates Obtained over a 73-Year Period. Plant Pathol. 2016, 65, 322–333. [Google Scholar] [CrossRef]
- Green, K.J.; Quintero-Ferrer, A.; Chikh-Ali, M.; Jones, R.A.C.; Karasev, A.V. Genetic Diversity of Nine Non-Recombinant Potato Virus y Isolates from Three Biological Strain Groups: Historical and Geographical Insights. Plant Dis. 2020, 104, 2317–2323. [Google Scholar] [CrossRef]
- Malmstrom, C.M.; Shu, R.; Linton, E.W.; Newton, L.A.; Cook, M.A. Barley Yellow Dwarf Viruses (BYDVs) Preserved in Herbarium Specimens Illuminate Historical Disease Ecology of Invasive and Native Grasses. J. Ecol. 2007, 95, 1153–1166. [Google Scholar] [CrossRef]
- Hartung, J.S.; Roy, A.; Fu, S.; Shao, J.; Schneider, W.L.; Brlansky, R.H. History and Diversity of Citrus Leprosis Virus Recorded in Herbarium Specimens. Phytopathology 2015, 105, 1277–1284. [Google Scholar] [CrossRef] [Green Version]
- Rieux, A.; Campos, P.; Duvermy, A.; Scussel, S.; Martin, D.; Gaudeul, M.; Lefeuvre, P.; Becker, N.; Lett, J.M. Contribution of Historical Herbarium Small RNAs to the Reconstruction of a Cassava Mosaic Geminivirus Evolutionary History. Sci. Rep. 2021, 11, 21280. [Google Scholar] [CrossRef]
- Jones, R.A.C.; Boonham, N.; Adams, I.P.; Fox, A. Historical Virus Isolate Collections: An Invaluable Resource Connecting Plant Virology’s Pre-Sequencing and Post-Sequencing Eras. Plant Pathol. 2021, 70, 235–248. [Google Scholar] [CrossRef]
- Ng, T.F.F.; Chen, L.F.; Zhou, Y.; Shapiro, B.; Stiller, M.; Heintzman, P.D.; Varsani, A.; Kondov, N.O.; Wong, W.; Deng, X.; et al. Preservation of Viral Genomes in 700-y-Old Caribou Feces from a Subarctic Ice Patch. Proc. Natl. Acad. Sci. USA 2014, 111, 16842–16847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-Thousand-Year-Old Distant Relative of Giant Icosahedral DNA Viruses with a Pandoravirus Morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 4274–4279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, M.; Lartigue, A.; Bertaux, L.; Jeudy, S.; Bartoli, J.; Lescot, M.; Alempic, J.M.; Ramus, C.; Bruley, C.; Labadie, K.; et al. In-Depth Study of Mollivirus Sibericum, a New 30,000-Yold Giant Virus Infecting Acanthamoeba. Proc. Natl. Acad. Sci. USA 2015, 112, E5327–E5335. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.P.; Tian, F.; Roux, S.; Gazitúa, M.C.; Solonenko, N.E.; Li, Y.F.; Davis, M.E.; Van Etten, J.L.; Mosley-Thompson, E.; Rich, V.I.; et al. Glacier Ice Archives Nearly 15,000-Year-Old Microbes and Phages. Microbiome 2021, 9, 160. [Google Scholar] [CrossRef]
- Jensen, T.Z.T.; Niemann, J.; Iversen, K.H.; Fotakis, A.K.; Gopalakrishnan, S.; Vågene, Å.J.; Pedersen, M.W.; Sinding, M.H.S.; Ellegaard, M.R.; Allentoft, M.E.; et al. A 5700 Year-Old Human Genome and Oral Microbiome from Chewed Birch Pitch. Nat. Commun. 2019, 10, 5520. [Google Scholar] [CrossRef] [Green Version]
- Duggan, A.T.; Klunk, J.; Porter, A.F.; Dhody, A.N.; Hicks, R.; Smith, G.L.; Humphreys, M.; McCollum, A.M.; Davidson, W.B.; Wilkins, K.; et al. The Origins and Genomic Diversity of American Civil War Era Smallpox Vaccine Strains. Genome Biol. 2020, 21, 175. [Google Scholar] [CrossRef]
- Brinkmann, A.; Souza, A.R.V.; Esparza, J.; Nitsche, A.; Damaso, C.R. Re-Assembly of Nineteenth-Century Smallpox Vaccine Genomes Reveals the Contemporaneous Use of Horsepox and Horsepox-Related Viruses in the USA. Genome Biol. 2020, 21, 4–9. [Google Scholar] [CrossRef]
- Duggan, A.T.; Holmes, E.C.; Poinar, H.N. Response to Brinkmann et al. Re-Assembly of 19th Century Smallpox Vaccine Genomes Reveals the Contemporaneous Use of Horsepox and Horsepox-Related Viruses in the United States. Genome Biol. 2020, 21, 20–21. [Google Scholar] [CrossRef]
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA Damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012567. [Google Scholar] [CrossRef]
- Orlando, L.; Allaby, R.; Skoglund, P.; Der Sarkissian, C.; Stockhammer, P.W.; Ávila-Arcos, M.C.; Fu, Q.; Krause, J.; Willerslev, E.; Stone, A.C.; et al. Ancient DNA Analysis. Nat. Rev. Methods Prim. 2021, 1, 14. [Google Scholar] [CrossRef]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.F.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of Damage in Genomic DNA Sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [Green Version]
- Allentoft, M.E.; Collins, M.; Harker, D.; Haile, J.; Oskam, C.L.; Hale, M.L.; Campos, P.F.; Samaniego, J.A.; Gilbert, T.P.M.; Willerslev, E.; et al. The Half-Life of DNA in Bone: Measuring Decay Kinetics in 158 Dated Fossils. Proc. R. Soc. B Biol. Sci. 2012, 279, 4724–4733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fordyce, S.L.; Kampmann, M.L.; van Doorn, N.L.; Gilbert, M.T.P. Long-Term RNA Persistence in Postmortem Contexts. Investig. Genet. 2013, 4, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höss, M.; Pääbo, S. DNA Extraction from Pleistocene Bones by a Silica-Based Purification Method. Nucleic Acids Res. 1993, 21, 3913–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirak, K.A.; Fernandes, D.M.; Cheronet, O.; Novak, M.; Gamarra, B.; Balassa, T.; Bernert, Z.; Cséki, A.; Dani, J.; Gallina, J.Z.; et al. A Minimally-Invasive Method for Sampling Human Petrous Bones from the Cranial Base for Ancient DNA Analysis. Biotechniques 2017, 62, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Rohland, N.; Hofreiter, M. Ancient Dna Extraction from Bones and Teeth. Nat. Protoc. 2007, 2, 1756–1762. [Google Scholar] [CrossRef] [Green Version]
- Briggs, A.W.; Stenzel, U.; Meyer, M.; Krause, J.; Kircher, M.; Pääbo, S. Removal of Deaminated Cytosines and Detection of in Vivo Methylation in Ancient DNA. Nucleic Acids Res. 2009, 38, e87. [Google Scholar] [CrossRef] [Green Version]
- Rohland, N.; Harney, E.; Mallick, S.; Nordenfelt, S.; Reich, D. Partial Uracil—DNA—Glycosylase Treatment for Screening of Ancient DNA. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20130624. [Google Scholar] [CrossRef]
- Beerenwinkel, N.; Zagordi, O. Ultra-Deep Sequencing for the Analysis of Viral Populations. Curr. Opin. Virol. 2011, 1, 413–418. [Google Scholar] [CrossRef]
- Lauring, A.S. Within-Host Viral Diversity: A Window into Viral Evolution. Annu. Rev. Virol. 2020, 7, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Beerenwinkel, N.; Günthard, H.F.; Roth, V.; Metzner, K.J. Challenges and Opportunities in Estimating Viral Genetic Diversity from Next-Generation Sequencing Data. Front. Microbiol. 2012, 3, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knapp, M.; Hofreiter, M. Next Generation Sequencing of Ancient DNA: Requirements, Strategies and Perspectives. Genes 2010, 1, 227–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun Metagenomics, from Sampling to Analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühlemann, B.; Vinner, L.; Margaryan, A.; Wilhelmson, H.; de la Fuente Castro, C.; Allentoft, M.E.; de Barros Damgaard, P.; Hansen, A.J.; Holtsmark Nielsen, S.; Strand, L.M.; et al. Diverse Variola Virus (Smallpox) Strains Were Widespread in Northern Europe in the Viking Age. Science 2020, 369, eaaw8977. [Google Scholar] [CrossRef]
- Guzmán-Solís, A.A.; Villa-Islas, V.; Bravo-López, M.J.; Sandoval-Velasco, M.; Wesp, J.K.; Gómez-Valdés, J.A.; de la Luz Moreno-Cabrera, M.; Meraz-Moreno, A.; Solís-Pichardo, G.; Schaaf, P.; et al. Ancient Viral Genomes Reveal Introduction of Human Pathogenic Viruses into Mexico during the Transatlantic Slave Trade. eLife 2021, 10, e68612. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid Adapter Trimming, Identification, and Read Merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Picard Toolkit. Available online: https://broadinstitute.github.io/picard/ (accessed on 19 April 2022).
- Bushnell, B. BBMap. Available online: http://sourceforge.net/projects/bbmap/ (accessed on 19 April 2022).
- Renaud, G.; Stenzel, U.; Kelso, J. LeeHom: Adaptor Trimming and Merging for Illumina Sequencing Reads. Nucleic Acids Res. 2014, 42, e141. [Google Scholar] [CrossRef]
- Mühlemann, B.; Margaryan, A.; De Barros Damgaard, P.; Allentoft, M.E.; Vinner, L.; Hansen, A.J.; Weber, A.; Bazaliiskii, V.I.; Molak, M.; Arneborg, J.; et al. Ancient Human Parvovirus B19 in Eurasia Reveals Its Long-Term Association with Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 7557–7562. [Google Scholar] [CrossRef] [Green Version]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borry, M.; Hübner, A.; Rohrlach, A.B.; Warinner, C. PyDamage: Automated Ancient Damage Identification and Estimation for Contigs in Ancient DNA de Novo Assembly. PeerJ 2021, 9, e11845. [Google Scholar] [CrossRef]
- Yue, Y.; Huang, H.; Qi, Z.; Dou, H.M.; Liu, X.Y.; Han, T.F.; Chen, Y.; Song, X.J.; Zhang, Y.H.; Tu, J. Evaluating Metagenomics Tools for Genome Binning with Real Metagenomic Datasets and CAMI Datasets. BMC Bioinform. 2020, 21, 334. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for Enhanced Metagenomic Taxonomic Profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef]
- Ye, S.H.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 2019, 178, 779–794. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; De Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning Metagenomic Contigs by Coverage and Composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef] [PubMed]
- Sedlar, K.; Kupkova, K.; Provaznik, I. Bioinformatics Strategies for Taxonomy Independent Binning and Visualization of Sequences in Shotgun Metagenomics. Comput. Struct. Biotechnol. J. 2017, 15, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Johansen, J.; Plichta, D.R.; Nissen, J.N.; Jespersen, M.L.; Shah, S.A.; Deng, L.; Stokholm, J.; Bisgaard, H.; Nielsen, D.S.; Sørensen, S.J.; et al. Genome Binning of Viral Entities from Bulk Metagenomics Data. Nat. Commun. 2022, 13, 965. [Google Scholar] [CrossRef] [PubMed]
- Arizmendi Cárdenas, Y.O.; Neuenschwander, S.; Malaspinas, A.-S. Benchmarking Metagenomics Classifiers on Ancient Viral DNA: A Simulation Study. PeerJ 2022, 10, e12784. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and Sensitive Classification of Metagenomic Sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Granehäll, L.; Huang, K.D.; Tett, A.; Manghi, P.; Paladin, A.; O’Sullivan, N.; Rota-Stabelli, O.; Segata, N.; Zink, A.; Maixner, F. Metagenomic Analysis of Ancient Dental Calculus Reveals Unexplored Diversity of Oral Archaeal Methanobrevibacter. Microbiome 2021, 9, 197. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Herbig, A.; Maixner, F.; Bos, K.I.; Zink, A.; Krause, J.; Huson, D.H. MALT: Fast Alignment and Analysis of Metagenomic DNA Sequence Data Applied to the Tyrolean Iceman. bioRxiv 2016, 050559. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN Analysis of Metagenomic Data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI Viral Genomes Resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, S.; Páez-Espino, D.; Chen, I.M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Reddy, T.; Nayfach, S.; Schulz, F.; Call, L.; et al. IMG/VR v3: An Integrated Ecological and Evolutionary Framework for Interrogating Genomes of Uncultivated Viruses. Nucleic Acids Res. 2021, 49, D764–D775. [Google Scholar] [CrossRef] [PubMed]
- Pratas, D.; Toppinen, M.; Pyoria, L.; Hedman, K.; Sajantila, A.; Perdomo, M.F. A Hybrid Pipeline for Reconstruction and Analysis of Viral Genomes at Multi-Organ Level. Gigascience 2020, 9, giaa086. [Google Scholar] [CrossRef]
- Schubert, M.; Ermini, L.; Der Sarkissian, C.; Jónsson, H.; Ginolhac, A.; Schaefer, R.; Martin, M.D.; Fernández, R.; Kircher, M.; McCue, M.; et al. Characterization of Ancient and Modern Genomes by SNP Detection and Phylogenomic and Metagenomic Analysis Using PALEOMIX. Nat. Protoc. 2014, 9, 1056–1082. [Google Scholar] [CrossRef]
- Pratas, D.; Hosseini, M.; Grilo, G.; Pinho, A.J.; Silva, R.M.; Caetano, T.; Carneiro, J.; Pereira, F. Metagenomic Composition Analysis of an Ancient Sequenced Polar Bear Jawbone from Svalbard. Genes 2018, 9, 445. [Google Scholar] [CrossRef] [Green Version]
- Fellows Yates, J.A.; Lamnidis, T.C.; Borry, M.; Valtueña, A.A.; Fagernäs, Z.; Clayton, S.; Garcia, M.U.; Neukamm, J.; Peltzer, A. Reproducible, Portable, and Efficient Ancient Genome Reconstruction with Nfcore/Eager. PeerJ 2021, 9, e10947. [Google Scholar] [CrossRef]
- Ingólfsson, Ó.; Wiig, Ø. Late Pleistocene Fossil Find in Svalbard: The Oldest Remains of a Polar Bear (Ursus Maritimus Phipps, 1744) Ever Discovered. Polar Res. 2009, 28, 455–462. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Ginolhac, A.; Rasmussen, M.; Gilbert, M.T.P.; Willerslev, E.; Orlando, L. MapDamage: Testing for Damage Patterns in Ancient DNA Sequences. Bioinformatics 2011, 27, 2153–2155. [Google Scholar] [CrossRef]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.F.; Orlando, L. MapDamage2.0: Fast Approximate Bayesian Estimates of Ancient DNA Damage Parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef]
- Skoglund, P.; Northoff, B.H.; Shunkov, M.V.; Derevianko, A.P.; Pääbo, S.; Krause, J.; Jakobsson, M. Separating Endogenous Ancient DNA from Modern Day Contamination in a Siberian Neandertal. Proc. Natl. Acad. Sci. USA 2014, 111, 2229–2234. [Google Scholar] [CrossRef] [Green Version]
- Neukamm, J.; Peltzer, A.; Nieselt, K. DamageProfiler: Fast Damage Pattern Calculation for Ancient DNA. Bioinformatics 2021, 37, 3652–3653. [Google Scholar] [CrossRef] [PubMed]
- Dinsdale, E.A.; Edwards, R.A.; Hall, D.; Angly, F.; Breitbart, M.; Brulc, J.M.; Furlan, M.; Desnues, C.; Haynes, M.; Li, L.; et al. Functional Metagenomic Profiling of Nine Biomes. Nature 2008, 452, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Eric Wommack, K.; Bhavsar, J.; Polson, S.W.; Chen, J.; Dumas, M.; Srinivasiah, S.; Furman, M.; Jamindar, S.; Nasko, D.J. VIROME: A Standard Operating Procedure for Analysis of Viral Metagenome Sequences. Stand. Genomic Sci. 2012, 6, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecuit, M.; Eloit, M. The Human Virome: New Tools and Concepts. Trends Microbiol. 2013, 21, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Bushman, F.D. The Human Virome: Assembly, Composition and Host Interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Abeles, S.R.; Robles-Sikisaka, R.; Ly, M.; Lum, A.G.; Salzman, J.; Boehm, T.K.; Pride, D.T. Human Oral Viruses Are Personal, Persistent and Gender-Consistent. ISME J. 2014, 8, 1753–1767. [Google Scholar] [CrossRef]
- Cadwell, K. Expanding the Role of the Virome: Commensalism in the Gut. J. Virol. 2015, 89, 1951–1953. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Härkönen, T.; Hämäläinen, A.M.; et al. Intestinal Virome Changes Precede Autoimmunity in Type I Diabetes-Susceptible Children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef] [Green Version]
- Nakatsu, G.; Zhou, H.; Wu, W.K.K.; Wong, S.H.; Coker, O.O.; Dai, Z.; Li, X.; Szeto, C.H.; Sugimura, N.; Lam, T.Y.T.; et al. Alterations in Enteric Virome Are Associated With Colorectal Cancer and Survival Outcomes. Gastroenterology 2018, 155, 529–541. [Google Scholar] [CrossRef] [Green Version]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Willner, D.; Furlan, M.; Haynes, M.; Schmieder, R.; Angly, F.E.; Silva, J.; Tammadoni, S.; Nosrat, B.; Conrad, D.; Rohwer, F. Metagenomic Analysis of Respiratory Tract DNA Viral Communities in Cystic Fibrosis and Non-Cystic Fibrosis Individuals. PLoS ONE 2009, 4, e7370. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Fornaciari, G.; Luciani, S.; Dowd, S.E.; Toranzos, G.A.; Marota, I.; Cano, R.J. Natural Mummification of the Human Gut Preserves Bacteriophage DNA. FEMS Microbiol. Lett. 2015, 363, fnv219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wibowo, M.C.; Yang, Z.; Borry, M.; Hübner, A.; Huang, K.D.; Tierney, B.T.; Zimmerman, S.; Barajas-Olmos, F.; Contreras-Cubas, C.; García-Ortiz, H.; et al. Reconstruction of Ancient Microbial Genomes from the Human Gut. Nature 2021, 594, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Rampelli, S.; Turroni, S.; Schnorr, S.L.; Soverini, M.; Quercia, S.; Barone, M.; Castagnetti, A.; Biagi, E.; Gallinella, G.; Brigidi, P.; et al. Characterization of the Human DNA Gut Virome across Populations with Different Subsistence Strategies and Geographical Origin. Environ. Microbiol. 2017, 19, 4728–4735. [Google Scholar] [CrossRef]
- Bédarida, S.; Dutour, O.; Buzhilova, A.P.; de Micco, P.; Biagini, P. Identification of Viral DNA (Anelloviridae) in a 200-Year-Old Dental Pulp Sample (Napoleon’s Great Army, Kaliningrad, 1812). Infect. Genet. Evol. 2011, 11, 358–362. [Google Scholar] [CrossRef]
- Holmes, E.C. Freezing Viruses in Time. Proc. Natl. Acad. Sci. USA 2014, 111, 16643–16644. [Google Scholar] [CrossRef] [Green Version]
- Patterson Ross, Z.; Klunk, J.; Fornaciari, G.; Giuffra, V.; Duchêne, S.; Duggan, A.T.; Poinar, D.; Douglas, M.W.; Eden, J.-S.; Holmes, E.C.; et al. The Paradox of HBV Evolution as Revealed from a 16th Century Mummy. PLoS Pathog. 2018, 14, e1006750. [Google Scholar] [CrossRef] [Green Version]
- Barquera, R.; Lamnidis, T.C.; Lankapalli, A.K.; Kocher, A.; Hernández-Zaragoza, D.I.; Nelson, E.A.; Zamora-Herrera, A.C.; Ramallo, P.; Bernal-Felipe, N.; Immel, A.; et al. Origin and Health Status of First-Generation Africans from Early Colonial Mexico. Curr. Biol. 2020, 30, 2078–2091.e11. [Google Scholar] [CrossRef]
- Neukamm, J.; Pfrengle, S.; Molak, M.; Seitz, A.; Francken, M.; Eppenberger, P.; Avanzi, C.; Reiter, E.; Urban, C.; Welte, B.; et al. 2000-Year-Old Pathogen Genomes Reconstructed from Metagenomic Analysis of Egyptian Mummified Individuals. BMC Biol. 2020, 18, 108. [Google Scholar] [CrossRef]
- Kocher, A.; Papac, L.; Barquera, R.; Key, F.M.; Spyrou, M.A.; Hübler, R.; Rohrlach, A.B.; Aron, F.; Stahl, R.; Wissgott, A.; et al. Ten Millennia of Hepatitis B Virus Evolution. Science 2021, 374, 182–188. [Google Scholar] [CrossRef]
- Gray, R.R.; Tanaka, Y.; Takebe, Y.; Magiorkinis, G.; Buskell, Z.; Seeff, L.; Alter, H.J.; Pybus, O.G. Evolutionary Analysis of Hepatitis C Virus Gene Sequences from 1953. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20130168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worobey, M.; Watts, T.D.; McKay, R.A.; Suchard, M.A.; Granade, T.; Teuwen, D.E.; Koblin, B.A.; Heneine, W.; Lemey, P.; Jaffe, H.W. 1970s and “Patient 0” HIV-1 Genomes Illuminate Early HIV/AIDS History in North America. Nature 2016, 539, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryseels, S.; Watts, T.D.; Mpolesha, J.M.K.; Larsen, B.B.; Lemey, P.; Muyembe-Tamfum, J.J.; Teuwen, D.E.; Worobey, M. A near Full-Length HIV-1 Genome from 1966 Recovered from Formalin-Fixed Paraffin-Embedded Tissue. Proc. Natl. Acad. Sci. USA 2020, 117, 12222–12229. [Google Scholar] [CrossRef] [PubMed]
- Fornaciari, G.; Zavaglia, K.; Giusti, L.; Vultaggio, C.; Ciranni, R. Human Papillomavirus in a 16th Century Mummy. Lancet 2003, 362, 1160. [Google Scholar] [CrossRef]
- Toppinen, M.; Perdomo, M.F.; Palo, J.U.; Simmonds, P.; Lycett, S.J.; Söderlund-Venermo, M.; Sajantila, A.; Hedman, K. Bones Hold the Key to DNA Virus History and Epidemiology. Sci. Rep. 2015, 5, 17226. [Google Scholar] [CrossRef] [Green Version]
- Li, H.C.; Fujiyoshi, T.; Lou, H.; Yashiki, S.; Sonoda, S.; Cartier, L.; Nunez, L.; Munoz, I.; Horai, S.; Tajima, K. The Presence of Ancient Human T-Cell Lymphotropic Virus Type I Provirus DNA in an Andean Mummy. Nat. Med. 1999, 5, 1428–1432. [Google Scholar] [CrossRef]
- Gessain, A.; Pecon-Slattery, J.; Meertens, L.; Mahieux, R. Origins of HTLV-1 in South America (Letter 1). Nat. Med. 2000, 6, 232. [Google Scholar] [CrossRef]
- Vandamme, A.-M.; Hall, W.W.; Lewis, M.J.; Goubau, P.; Salemi, M. Origins of HTLV-1 in South America (Letter 2). Nat. Med. 2000, 6, 232–233. [Google Scholar] [CrossRef]
- Reid, A.H.; Fanning, T.G.; Hultin, J.V.; Taubenberger, J.K. Origin and Evolution of the 1918 “Spanish” Influenza Virus Hemagglutinin Gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1651–1656. [Google Scholar] [CrossRef] [Green Version]
- Reid, A.H.; Fanning, T.G.; Janczewski, T.A.; Taubenberger, J.K. Characterization of the 1918 “Spanish” Influenza Virus Neuraminidase Gene. Proc. Natl. Acad. Sci. USA 2000, 97, 6785–6790. [Google Scholar] [CrossRef] [Green Version]
- Basler, C.F.; Reid, A.H.; Dybing, J.K.; Janczewski, T.A.; Fanning, T.G.; Zheng, H.; Salvatore, M.; Perdue, M.L.; Swayne, D.E.; García-Sastre, A.; et al. Sequence of the 1918 Pandemic Influenza Virus Nonstructural Gene (NS) Segment and Characterization of Recombinant Viruses Bearing the 1918 NS Genes. Proc. Natl. Acad. Sci. USA 2001, 98, 2746–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, A.H.; Fanning, T.G.; Janczewski, T.A.; McCall, S.; Taubenberger, J.K. Characterization of the 1918 “Spanish” Influenza Virus Matrix Gene Segment. J. Virol. 2002, 76, 10717–10723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, A.H.; Fanning, T.G.; Janczewski, T.A.; Lourens, R.M.; Taubenberger, J.K. Novel Origin of the 1918 Pandemic Influenza Virus Nucleoprotein Gene. J. Virol. 2004, 78, 12462–12470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubenberger, J.K.; Reid, A.H.; Lourens, R.M.; Wang, R.; Jin, G.; Fanning, T.G. Characterization of the 1918 Influenza Virus Polymerase Genes. Nature 2005, 437, 889–893. [Google Scholar] [CrossRef]
- Xiao, Y.L.; Kash, J.C.; Beres, S.B.; Sheng, Z.M.; Musser, J.M.; Taubenberger, J.K. High-Throughput RNA Sequencing of a Formalin-Fixed, Paraffin-Embedded Autopsy Lung Tissue Sample from the 1918 Influenza Pandemic. J. Pathol. 2013, 229, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Düx, A.; Lequime, S.; Patrono, L.V.; Vrancken, B.; Boral, S.; Gogarten, J.F.; Hilbig, A.; Horst, D.; Merkel, K.; Prepoint, B.; et al. Measles Virus and Rinderpest Virus Divergence Dated to the Sixth Century BCE. Science 2020, 368, 1367–1370. [Google Scholar] [CrossRef]
- Larsen, B.B.; Cole, K.L.; Worobey, M. Ancient DNA Provides Evidence of 27,000-Year-Old Papillomavirus Infection and Long-Term Codivergence with Rodents. Virus Evol. 2018, 4, vey014. [Google Scholar] [CrossRef]
- Calvignac, S.; Terme, J.M.; Hensley, S.M.; Jalinot, P.; Greenwood, A.D.; Hänni, C. Ancient DNA Identification of Early 20th Century Simian T-Cell Leukemia Virus Type 1. Mol. Biol. Evol. 2008, 25, 1093–1098. [Google Scholar] [CrossRef]
- Castello, J.D.; Rogers, S.O.; Starmer, W.T.; Catranis, C.M.; Ma, L.; Bachand, G.D.; Zhao, Y.; Smith, J.E. Detection of Tomato Mosaic Tobamovirus RNA in Ancient Glacial Ice. Polar Biol. 1999, 22, 207–212. [Google Scholar] [CrossRef]
- Biagini, P.; Thèves, C.; Balaresque, P.; Géraut, A.; Cannet, C.; Keyser, C.; Nikolaeva, D.; Gérard, P.; Duchesne, S.; Orlando, L.; et al. Variola Virus in a 300-Year-Old Siberian Mummy. N. Engl. J. Med. 2012, 367, 2057–2059. [Google Scholar] [CrossRef] [Green Version]
- Meffray, A.; Ardagna, Y.; Sillano, B.; Parmentier, S.; Pouget, B.; Signoli, M.; Biagini, P. Variola Virus DNA in Skeletal Remains, 17th to 18th Centuries, Southeastern France. Clin. Microbiol. Infect. 2021, 27, 1871–1872. [Google Scholar] [CrossRef] [PubMed]
- Duggan, A.T.; Perdomo, M.F.; Piombino-Mascali, D.; Marciniak, S.; Poinar, D.; Emery, M.V.; Buchmann, J.P.; Duchêne, S.; Jankauskas, R.; Humphreys, M.; et al. 17th Century Variola Virus Reveals the Recent History of Smallpox. Curr. Biol. 2016, 26, 3407–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smithson, C.; Imbery, J.; Upton, C. Re-Assembly and Analysis of an Ancient Variola Virus Genome. Viruses 2017, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Pajer, P.; Dresler, J.; Kabíckova, H.; Písa, L.; Aganov, P.; Fucik, K.; Elleder, D.; Hron, T.; Kuzelka, V.; Velemínsky, P.; et al. Characterization of Two Historic Smallpox Specimens from a Czech Museum. Viruses 2017, 9, 200. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.F.; Duggan, A.T.; Poinar, H.N.; Holmes, E.C. Comment: Characterization of Two Historic Smallpox Specimens from a Czech Museum. Viruses 2017, 9, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, G.; Neukamm, J.; Baalsrud, H.T.; Breidenstein, A.M.; Ravinet, M.; Phillips, C.; Rühli, F.; Bouwman, A.; Schuenemann, V.J. Variola Virus Genome Sequenced from an Eighteenth-Century Museum Specimen Supports the Recent Origin of Smallpox: 18th Century Variola Virus Genome. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190572. [Google Scholar] [CrossRef]
- Babkin, I.V.; Babkina, I.N.; Tikunova, N.V. An Update of Orthopoxvirus Molecular Evolution. Viruses 2022, 14, 388. [Google Scholar] [CrossRef]
- Tumpey, T.M.; Basler, C.F.; Aguilar, V.P.; Basler, C.F.; Aguilar, P.V.; Zeng, H.; Solórzano, A.; Swayne, D.E.; Cox, N.J.; Katz, J.M.; et al. Characterization of the Reconstructed 1918 Spanish Influenza Pandemic Virus. Science 2005, 310, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, M.S. Measles Periodicity and Community Size. J. R. Stat. Soc. Ser. A 1957, 120, 48. [Google Scholar] [CrossRef]
- Black, F.L. Measles Endemicity in Insular Populations: Critical Community Size and Its Evolutionary Implication. J. Theor. Biol. 1966, 11, 207–211. [Google Scholar] [CrossRef]
- Keeling, M.J.; Grenfell, B.T. Disease Extinction and Community Size: Modeling the Persistence of Measles. Science 1997, 275, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Littlejohn, M.; Locarnini, S.; Yuen, L. Origins and Evolution of Hepatitis B Virus and Hepatitis D Virus. Cold Spring Harb. Perspect. Med. 2016, 6, a021360. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Holmes, E.C. Bayesian Estimates of the Evolutionary Rate and Age of Hepatitis B Virus. J. Mol. Evol. 2007, 65, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraskevis, D.; Angelis, K.; Magiorkinis, G.; Kostaki, E.; Ho, S.Y.W.; Hatzakis, A. Dating the Origin of Hepatitis B Virus Reveals Higher Substitution Rate and Adaptation on the Branch Leading to F/H Genotypes. Mol. Phylogenet. Evol. 2015, 93, 44–54. [Google Scholar] [CrossRef]
- Zehender, G.; Ebranati, E.; Gabanelli, E.; Sorrentino, C.; Presti, A.L.; Tanzi, E.; Ciccozzi, M.; Galli, M. Enigmatic Origin of Hepatitis B Virus: An Ancient Travelling Companion or a Recent Encounter? World J. Gastroenterol. 2014, 20, 7622–7634. [Google Scholar] [CrossRef]
- Ho, S.Y.W.; Duchêne, S.; Molak, M.; Shapiro, B. Time-Dependent Estimates of Molecular Evolutionary Rates: Evidence and Causes. Mol. Ecol. 2015, 24, 6007–6012. [Google Scholar] [CrossRef]
- Firth, C.; Kitchen, A.; Shapiro, B.; Suchard, M.A.; Holmes, E.C.; Rambaut, A. Using Time-Structured Data to Estimate Evolutionary Rates of Double-Stranded DNA Viruses. Mol. Biol. Evol. 2010, 27, 2038–2051. [Google Scholar] [CrossRef] [Green Version]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of Viral Mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [Green Version]
- Perelson, A.S. Modelling Viral and Immune System Dynamics. Nat. Rev. Immunol. 2002, 2, 28–36. [Google Scholar] [CrossRef]
- Irwin, K.K.; Renzette, N.; Kowalik, T.F.; Jensen, J.D. Antiviral Drug Resistance as an Adaptive Process. Virus Evol. 2016, 2, vew014. [Google Scholar] [CrossRef] [Green Version]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Gojobori, T.; Moriyama, E.N.; Kimura, M. Molecular Clock of Viral Evolution, and the Neutral Theory. Proc. Natl. Acad. Sci. USA 1990, 87, 10015–10018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, G.M.; Rambaut, A.; Pybus, O.G.; Holmes, E.C. Rates of Molecular Evolution in RNA Viruses: A Quantitative Phylogenetic Analysis. J. Mol. Evol. 2002, 54, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R. From Molecular Genetics to Phylodynamics: Evolutionary Relevance of Mutation Rates across Viruses. PLoS Pathog. 2012, 8, e1002685. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of Evolutionary Change in Viruses: Patterns and Determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Kosakovsky Pond, S.L. Purifying Selection Can Obscure the Ancient Age of Viral Lineages. Mol. Biol. Evol. 2011, 28, 3355–3365. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.Y.W.; Lanfear, R.; Bromham, L.; Phillips, M.J.; Soubrier, J.; Rodrigo, A.G.; Cooper, A. Time-Dependent Rates of Molecular Evolution. Mol. Ecol. 2011, 20, 3087–3101. [Google Scholar] [CrossRef]
- Duchêne, S.; Holmes, E.C.; Ho, S.Y.W. Analyses of Evolutionary Dynamics in Viruses Are Hindered by a Time-Dependent Bias in Rate Estimates. Proc. R. Soc. B Biol. Sci. 2014, 281, 20140732. [Google Scholar] [CrossRef] [Green Version]
- Aiewsakun, P.; Katzourakis, A. Time-Dependent Rate Phenomenon in Viruses. J. Virol. 2016, 90, 7184–7195. [Google Scholar] [CrossRef] [Green Version]
- Ghafari, M.; Simmonds, P.; Pybus, O.G.; Katzourakis, A. A Mechanistic Evolutionary Model Explains the Time-Dependent Pattern of Substitution Rates in Viruses. Curr. Biol. 2021, 31, 4689–4696. [Google Scholar] [CrossRef]
- Lythgoe, K.A.; Gardner, A.; Pybus, O.G.; Grove, J. Short-Sighted Virus Evolution and a Germline Hypothesis for Chronic Viral Infections. Trends Microbiol. 2017, 25, 336–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, A.; Lemey, P.; Hurles, M.; Moyes, C.; Horn, S.; Pryor, J.; Malani, J.; Supuri, M.; Masta, A.; Teriboriki, B.; et al. Genomic Analysis of Hepatitis B Virus Reveals Antigen State and Genotype as Sources of Evolutionary Rate Variation. Viruses 2011, 3, 83–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-Y.; Liu, C.; Chien, W.-H.; Wu, L.-L.; Tao, Y.; Wu, D.; Lu, X.; Hsieh, C.-H.; Chen, P.-J.; Wang, H.-Y.; et al. New Insights into the Evolutionary Rate of Hepatitis B Virus at Different Biological Scales. J. Virol. 2015, 89, 3512–3522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lythgoe, K.A.; Fraser, C. New Insights into the Evolutionary Rate of HIV-1 at the within-Host and Epidemiological Levels. Proc. R. Soc. B Biol. Sci. 2012, 279, 3367–3375. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Katzourakis, A. Time Dependency of Foamy Virus Evolutionary Rate Estimates. BMC Evol. Biol. 2015, 15, 119. [Google Scholar] [CrossRef] [Green Version]
- Holmes, E.C.; Duchêne, S. Evolutionary Stasis of Viruses? Nat. Rev. Microbiol. 2019, 17, 329. [Google Scholar] [CrossRef]
- Emerson, B.C.; Hickerson, M.J. Lack of Support for the Time-Dependent Molecular Evolution Hypothesis. Mol. Ecol. 2015, 24, 702–709. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Bos, K.I.; Kühnert, D.; Herbig, A.; Esquivel-Gomez, L.R.; Andrades Valtueña, A.; Barquera, R.; Giffin, K.; Kumar Lankapalli, A.; Nelson, E.A.; Sabin, S.; et al. Paleomicrobiology: Diagnosis and Evolution of Ancient Pathogens. Annu. Rev. Microbiol. 2019, 73, 639–666. [Google Scholar] [CrossRef]
- Tong, K.J.; Duchêne, D.A.; Duchêne, S.; Geoghegan, J.L.; Ho, S.Y.W. A Comparison of Methods for Estimating Substitution Rates from Ancient DNA Sequence Data. BMC Evol. Biol. 2018, 18, 70. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Pybus, O.G.; Rambaut, A.; Forsberg, R.; Rodrigo, A.G. Measurably Evolving Populations. Trends Ecol. Evol. 2003, 18, 481–488. [Google Scholar] [CrossRef]
- Korber, B.; Muldoon, M.; Theiler, J.; Gao, F.; Gupta, R.; Lapedes, A.; Hahn, B.H.; Wolinsky, S.; Bhattacharya, T. Timing the Ancestor of the HIV-1 Pandemic Strains. Science 2000, 288, 1789–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieux, A.; Balloux, F. Inferences from Tip-Calibrated Phylogenies: A Review and a Practical Guide. Mol. Ecol. 2016, 25, 1911–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchêne, S.; Duchêne, D.; Holmes, E.C.; Ho, S.Y.W. The Performance of the Date-Randomization Test in Phylogenetic Analyses of Time-Structured Virus Data. Mol. Biol. Evol. 2015, 32, 1895–1906. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining Viral Signal from Microbial Genomic Data. PeerJ 2015, 2015, e985. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef]
- Amgarten, D.; Braga, L.P.P.; da Silva, A.M.; Setubal, J.C. MARVEL, a Tool for Prediction of Bacteriophage Sequences in Metagenomic Bins. Front. Genet. 2018, 9, 304. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A Novel k-Mer Based Tool for Identifying Viral Sequences from Assembled Metagenomic Data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of Huge Phages from across Earth’s Ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, R.; Nishimura, L.; Nguyen, P.T.; Ito, J.; Parrish, N.F.; Mori, H.; Kurokawa, K.; Nakaoka, H.; Inoue, I. Comprehensive Discovery of CRISPR-Targeted Terminally Redundant Sequences in the Human Gut Metagenome: Viruses, Plasmids, and More. PLoS Comput. Biol. 2021, 17, e1009428. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short Motif Sequences Determine the Targets of the Prokaryotic CRISPR Defence System. Microbiology 2009, 155, 733–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
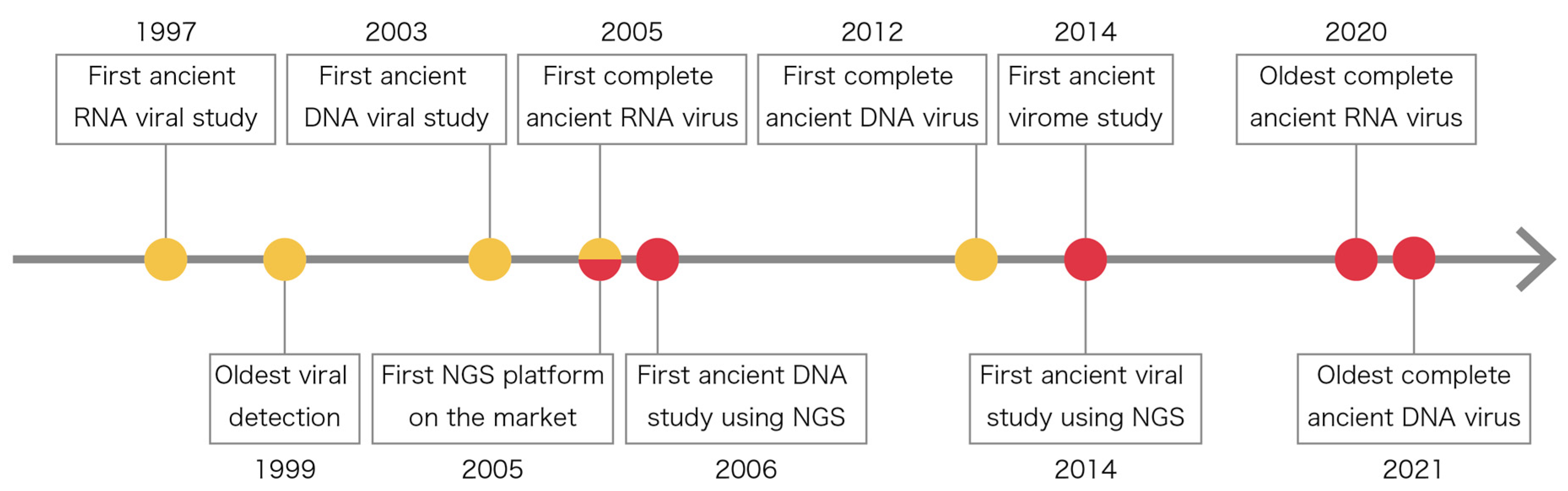

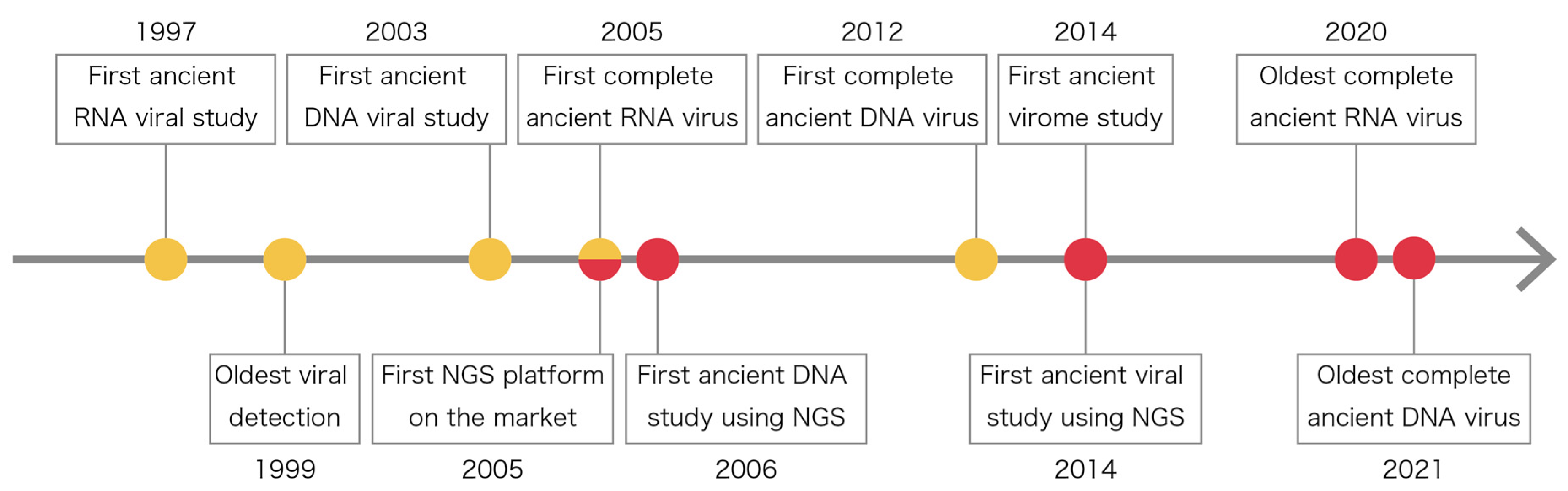{kind=link}
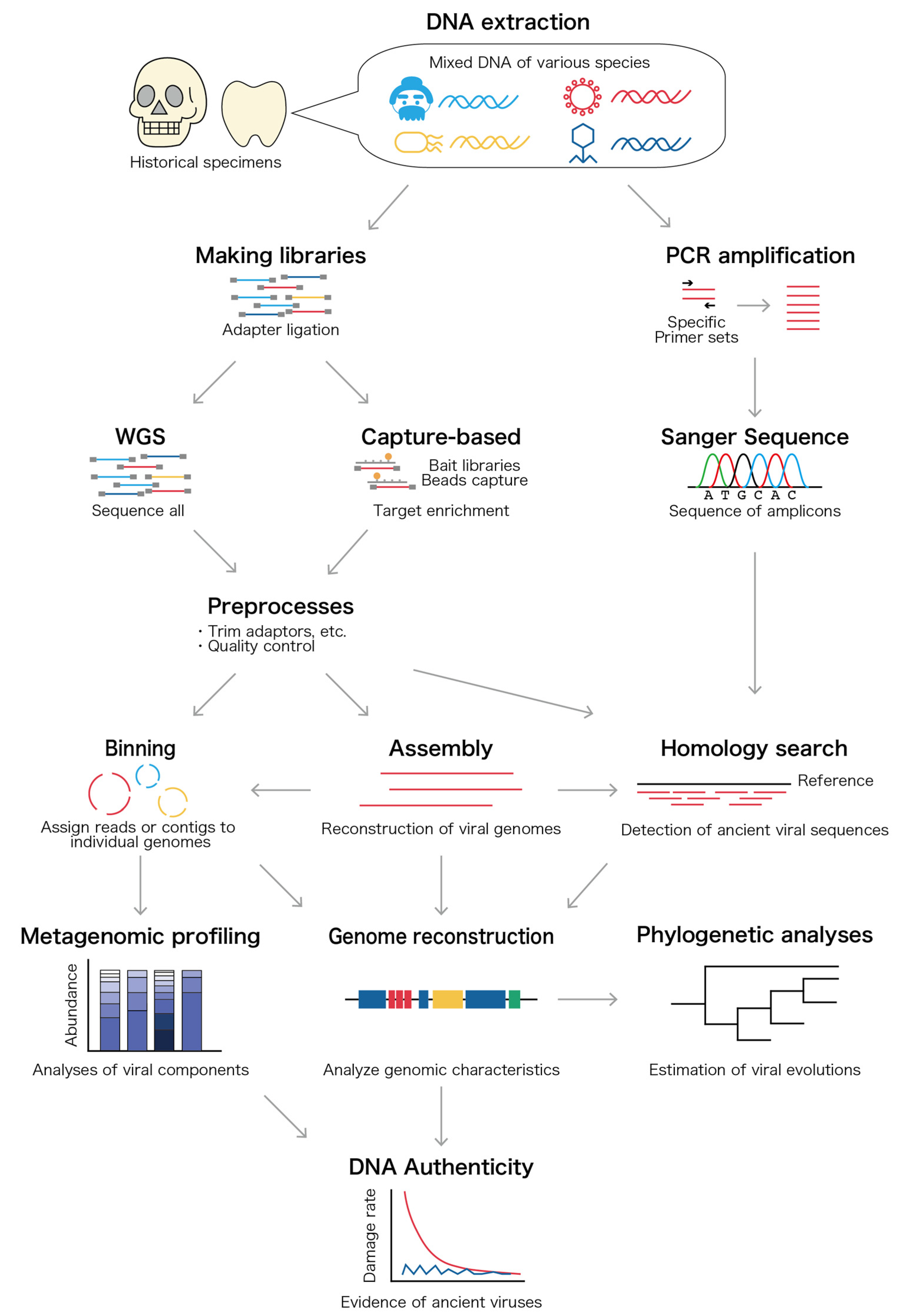{kind=link}
| Species Name | Type | Host | Reference Accession ID | Reference Length (kb) | Detected Length (kb) 1 | Method | Sample | Sample Age (ya) 2 | Region | Accession ID of Ancient Viral Genomes | Accession ID of Raw Reads | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| African cassava mosaic virus (ACMV) | ssDNA 3 | Manihot glaziovii | NC_001467, NC_001468 | 5.5 | 5.5 | PCR 10, WGS 11 | Leaf of Manihot glaziovii specimen | 94 | Bambari, Central African Republic | MW788219, MW788220 | PRJNA698751 | Rieux et al., 2021 [41] |
| Anelloviridae | ssDNA | Homo sapiens | AB303563 | 3.2 | 0.078 | PCR | Dental pulp | 200 | Kaliningrad, Russia | NA | NA | Bédarida et al., 2011 [128] |
| Ancient caribou feces-associated virus (aCFV) | DNA | Plant | NA | NA | 2.2 | PCR | Coprolite | 700 | Northwest Territories, Canada | KJ938716 | NA | Ng et al., 2014 [43], Holmes, 2014 [129] |
| Ancient Northwest Territories cripavirus (aNCV) | +ssRNA 4 | Insect | NA | NA | 1.8 | PCR | Coprolite | 700 | Northwest Territories, Canada | KJ938718 | NA | Ng et al., 2014 [43], Holmes, 2014 [129] |
| Barely stripe mosaic virus (BSMV) | +ssRNA | Horedum vulgare | NC_003469, NC_003481, NC_003478 | 10.2 | 10.2 | WGS | Barely grain | 750 | Upper Nubia, Egypt | NA | NA | Smith et al., 2014 [29] |
| Barely yellow dwarf virus (BYDV) | +ssRNA | Avena fatua, Danthonia californica, Glyceria elata, Koeleria macrantha, Phalaris coerulescens | NC_004750 | 5.7 | 3.6 | RT-PCR 12 | Herbarium specimens | 82–105 | US | DQ115532–DQ115534, DQ118372, DQ631844–DQ631846, DQ631856, DQ631857 | NA | Malmstrom et al., 2007 [39] |
| Citrus leprosis virus (CiLV) | +ssRNA | Citrus aurantium, Citrus sinensis | NC_008169, NC_008170 | 13.7 | 12.7 | WGS | Herbarium specimens | 55–90 | US, Mexico, Argentina, Brazil | KT187687–KT187693 | NA | Hartung et al., 2015 [40] |
| Epstein–Barr virus (EBV) | dsDNA | Homo sapiens | NC_007605 | 171.8 | 22.9 | WGS | Chewed birch pitch | 5700 | Island of Lolland, Denmark | NA | PRJEB30280 | Jensen et al., 2019 [47] |
| Hepatitis B virus (HBV) | dsDNA-RT 5 | Homo sapiens | NC_001611 | 3.2 | 3.2 | PCR | Liver of a mummy | 500 | Yangju, Korea | JN315779 | NA | Kahila Bar-Gal et al., 2012 [33] |
| 3.2 | Capture 13 | Distal femur, skin, muscle of a mummy | 500 | Naples, Italy | MG585269 | NA | Patterson et al., 2018 [130] | |||||
| 3.2 | WGS, Capture | Tooth cementum, petrous bones | 822–4488 | Central to western Eurasia | ERS2295383–ERS2295394 | PRJEB9021, PRJEB20658 | Mühlemann et al., 2018 [14] | |||||
| 3.2 | WGS | Teeth | 340–5000 | Germany | NA | PRJEB24921 | Krause-Kyora et al., 2018 [15] | |||||
| 2.9 | WGS, Capture | Tooth | 396–569 | Mexico City, Mexico | NA | PRJEB37490 | Barquera, et al., 2020 [131] | |||||
| 3.1 | WGS | Soft tissue and bone of a mummy | 2000 | Abusir el-Meleq, Egypt | NA | PRJEB33848 | Neukamm et al., 2020 [132] | |||||
| 3.2 | WGS, Capture | Tooth root | 500 | Mexico City, Mexico | MT108214 | Available at Dryad 14 | Guzmán-Solís et al., 2021 [67] | |||||
| 3.2 | WGS, Capture | Teeth, bones, petrous bones | 400–10,500 | Eurasia and US | ERS6597748–ERS6597884 | PRJEB45699 | Kocher et al., 2021 [133] | |||||
| Hepatitis C virus (HCV) | +ssRNA | Homo sapiens | NC_004102 | 9.7 | 0.336 | RT-PCR | Archived blood samples | 69 | US | KF261594, KF261595 | NA | Gray et al., 2013 [134] |
| Human immunodeficiency virus type 1 (HIV-1) | ssRNA-RT 6 | Homo sapiens | NC_001802 | 9.2 | ~0.3 | RT-PCR | Plasma samples | 63 | Kinshasa, Democratic Republic of Congo | NA | NA | Zhu et al., 1998 [26] |
| 8.6 | RT-PCR | Frozen serum samples | 50 | New York City, US | KJ704787–KJ704797 | NA | Worobey et al., 2016 [135] | |||||
| 8.3 | RT-PCR, amplicon sequence | Formalin-fixed paraffin-embedded tissues | 56 | Kinshasa, Democratic Republic of Congo | MN082768 | NA | Gryseels et al., 2020 [136] | |||||
| Human papillomavirus (HPV) | dsDNA 7 | Homo sapiens | NC_027779 | 7.3 | 0.141 | PCR | Mummy of a Renaissance noble woman | 454 | Naples, Italy | NA | NA | Fornaciar et al., 2003 [137] |
| Human parvovirus B19 (B19V) | ssDNA | Homo sapiens | NC_000883 | 5.6 | 0.275 | PCR | Long bones | 92 | Karelia district, Finland | NA | NA | Toppinen et al., 2015 [138] |
| 5.9 | WGS | Dental, skeletal remains | 500–6900 | Eurasia, Southeast Asia, Greenland | NA | PRJEB26712 15 | Mühlemann et al., 2018 [73] | |||||
| 4.4 | WGS, Capture | Tooth roots | 500 | Mexico City, Mexico | MT108215–MT108217 | Available at Dryad 14 | Guzmán-Solís et al., 2021 [67] | |||||
| Human T-cell leukemia virus type 1 (HTLV-1) | ssRNA-RT | Homo sapiens | NC_001436 | 8.5 | 0.316 | PCR | Mummy | 500 | Andean, US | NA | NA | Li et al., 1999 [139], Gessain et al., 2000 [140], Vandamme et al., 2000 [141] |
| Influenza A virus | -ssRNA 8 | Homo sapiens | NC_026431-NC_026438 | 13.2 | 12.7 | RT-PCR | Formalin-fixed paraffin-embedded lung tissues | 104 | US | AF116575, AF250356, AF333238, AY130766, AY744935, DQ208309–DQ208311 | NA | Taubenberger et al., 1997 [12], Reid et al., 1999 [142], Reid et al., 2000 [143], Basler et al., 2001 [144], Reid et al., 2002 [145], Reid et al., 2004 [146], Taubenberger et al., 2005 [147] |
| 12.7 | RT-PCR, WGS | Formalin-fixed paraffin-embedded lung tissues | 104 | New York City, US | NA | PRJNA178740 | Xiao et al., 2013 [148] | |||||
| Measles morbillivirus (MeV) | -ssRNA | Homo sapiens | NC_001498 | 15.8 | 15.8 | WGS | Formalin-fixed paraffin-embedded lung tissues | 110 | Berlin, Germany | NA | PRJEB36265 | Düx et al., 2020 [149] |
| Mollivirus sibericum | dsDNA | Acanthamoeba castellanii | NA | NA | 651 | WGS | Permafrost layer | 30,000 | Northeast Siberia, Russia | KR921745 | NA | Legendre et al., 2015 [45] |
| Papillomavirus | dsDNA | Neuroma cinera | MF416381 | 7.4 | 0.677 | PCR | Unwashed midden materials | 27,000 | Arizona, US | MH136586, MH136587 | NA | Larsen et al., 2018 [150] |
| Pithovirus sibericum | dsDNA | Acanthamoeba castellanii | NA | NA | 610 | WGS | Permafrost layer | 30,000 | Northeast Siberia, Russia | KF740664 | NA | Legendre et al., 2014 [44] |
| Potato virus X (PVX) | +ssRNA | Solanum tuberosum | NC_011620 | 6.4 | 0.75 | RT-PCR | Freeze dried leaves | 38–52 | Australia, England | GU384732–GU384734, GU384737–GU384738 | NA | Cox and Jones et al., 2010 [35] |
| Potato virus Y (PVY) | +ssRNA | Solanum tuberosum | NC_001616 | 9.7 | 9.7 | RT-PCR | Potato | 84 | Netherlands | EU563512 | NA | Dullemans et al., 2011 [36] |
| 9.7 | WGS | Freeze-died PVY cultures | 38–79 | UK | KP691317–KP691330, MT200665–MT200668 | NA | Kehoe and Jones, 2016 [37], Green et al., 2020 [38] | |||||
| Simian T-lymphotropic virus type 1 (STLV-1) | ssRNA-RNA | Cercopithecus aethiops | MF622054 | 8.4 | 0.467 | PCR | Skeletons | 122 | Central Africa | NA | NA | Calvignac et al., 2008 [151] |
| Siphovirus contig89 (CT89) | dsDNA | Schalia meyeri | KF594184 | 2.4 | 4.2 | WGS | Dental pulp | 3800 | Hokkaido, Japan | LC585292 | PRJDB7235 | Nishimura et al., 2021 [30] |
| Tomato mosaic tobamovirus (ToMV) | +ssRNA | Dicotyledonous, monocotyledonous | NC_002692 | 6.4 | 0.347 | RT-PCR | Ice cores | <500–140,000 | Greenland | NA | NA | Castello et al., 1999 [152] |
| Variola virus (VARV) | dsDNA | Homo sapiens | NC_001611 | 185.6 | 0.718 | PCR | Pulmonary tissue of a mummy | 300 | Siberia, Russia | JX080525–JX080527 | NA | Biagini et al., 2012 [153] |
| 0.43 | Skeleton | 300 | Marseille city, France | NA | NA | Meffray et al., 2021 [154] | ||||||
| 166.8 | Capture | Soft tissue of a mummy | 367–379 | Vilnius, Lithuania | KY358055, BK010317 | PRJNA348754 | Duggan et al., 2016 [155], Smithson et al., 2017 [156] | |||||
| 185.4 | WGS | Forefoot and piece of skin | 100 | Prague, Czech | LT706528, LT706529 | PRJEB18730 | Pajer et al., 2017 [157], Porter et al., 2017 [158] | |||||
| 158.1 | Ethanol-fixed infant leg | 229–262 | London, England | NA | PRJEB35140 | Ferrari et al., 2020 [159] | ||||||
| 192.3 | WGS, Capture | Skeletons | 970–1400 | Northern Europe | LR800244–LR800247 | PRJEB38129 | Mühlemann et al., 2020 [66], Babkin et al., 2022 [160] | |||||
| Vaccinia virus (VACV) | dsDNA | Homo sapiens | M35027 | 191.7 | 184.7 | WGS, Capture | Vaccination kits | 156 | Philadelphia, US | MN369532 | PRJNA561155 | Duggan et al., 2020 [48], Brinkmann, et al., 2020 [49], Duggan et al., 2020 [50] |
| Zea may chrysovirus 1 (ZMCV1) | dsRNA 9 | Zea mays | NA | NA | 11.3 | WGS, RT-PCR | Maize cobs | 1000 | Antelope house, US | MH931189–MH931208, MH936006, MH936007, MH936014–MH936017 | NA | Peyambari et al., 2019 [34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishimura, L.; Fujito, N.; Sugimoto, R.; Inoue, I. Detection of Ancient Viruses and Long-Term Viral Evolution. Viruses 2022, 14, 1336. https://doi.org/10.3390/v14061336
Nishimura L, Fujito N, Sugimoto R, Inoue I. Detection of Ancient Viruses and Long-Term Viral Evolution. Viruses. 2022; 14(6):1336. https://doi.org/10.3390/v14061336
Chicago/Turabian StyleNishimura, Luca, Naoko Fujito, Ryota Sugimoto, and Ituro Inoue. 2022. "Detection of Ancient Viruses and Long-Term Viral Evolution" Viruses 14, no. 6: 1336. https://doi.org/10.3390/v14061336
APA StyleNishimura, L., Fujito, N., Sugimoto, R., & Inoue, I. (2022). Detection of Ancient Viruses and Long-Term Viral Evolution. Viruses, 14(6), 1336. https://doi.org/10.3390/v14061336