
Development of a Singleplex Real-Time Reverse Transcriptase PCR Assay for Pan-Dengue Virus Detection and Quantification

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of In Vitro RNA Standards
2.2. Preparation of Viruses from Cultured Cells
2.3. Clinical Specimens
2.4. Primers/Probes Design
2.5. RNA Extraction and Quantitative Real-Time RT-PCR
2.6. Statistical Analysis
3. Results
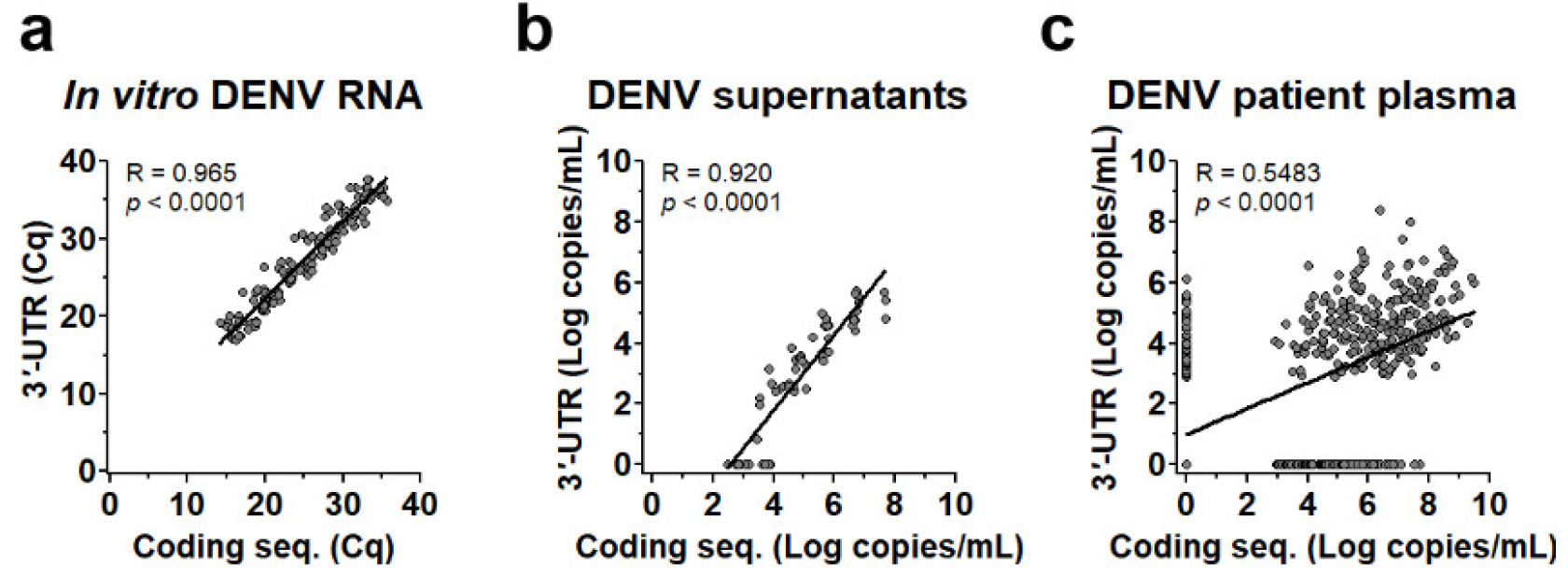
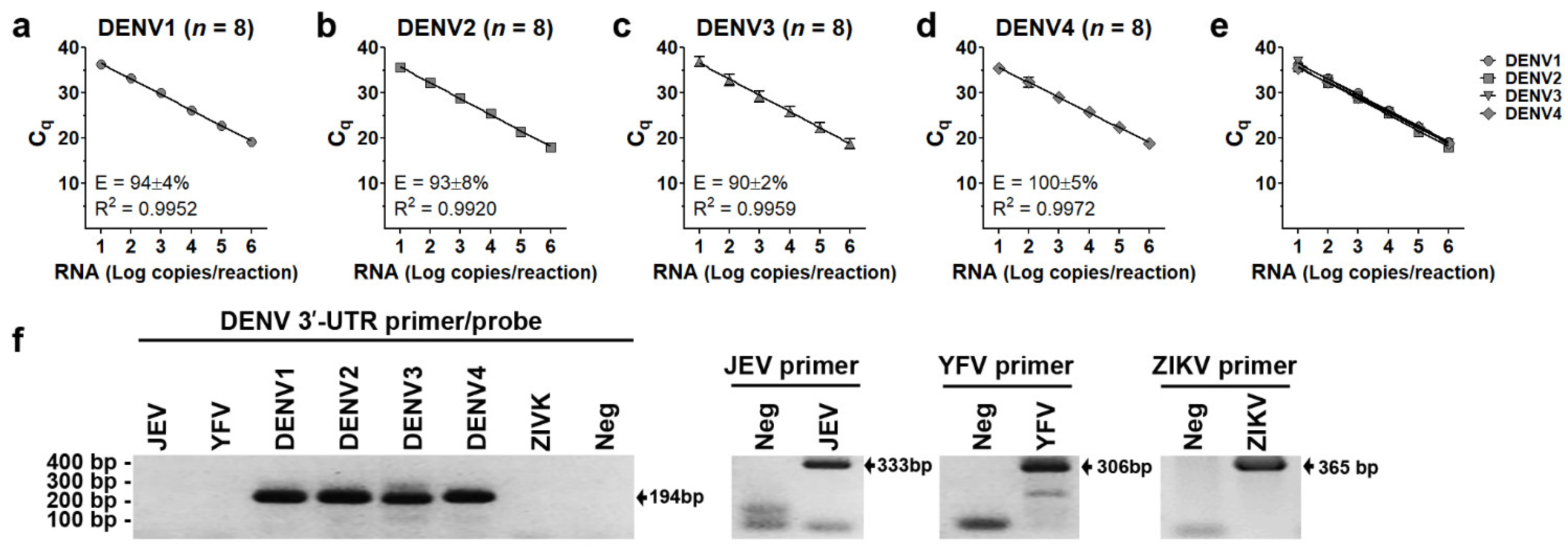
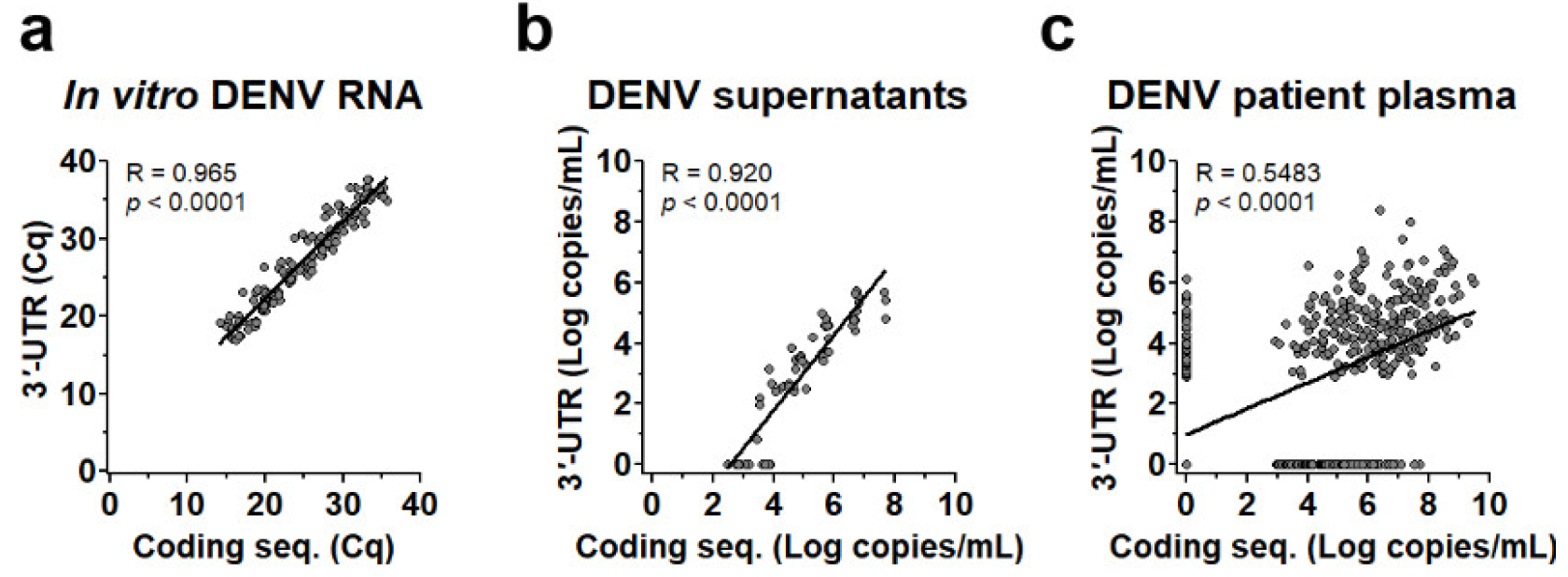
3.1. Performance of 3′-UTR Real-Time RT-PCR Assay
3.2. Efficiency of DENV Viral Genome Quantification of 3′-UTR Primers/Probes
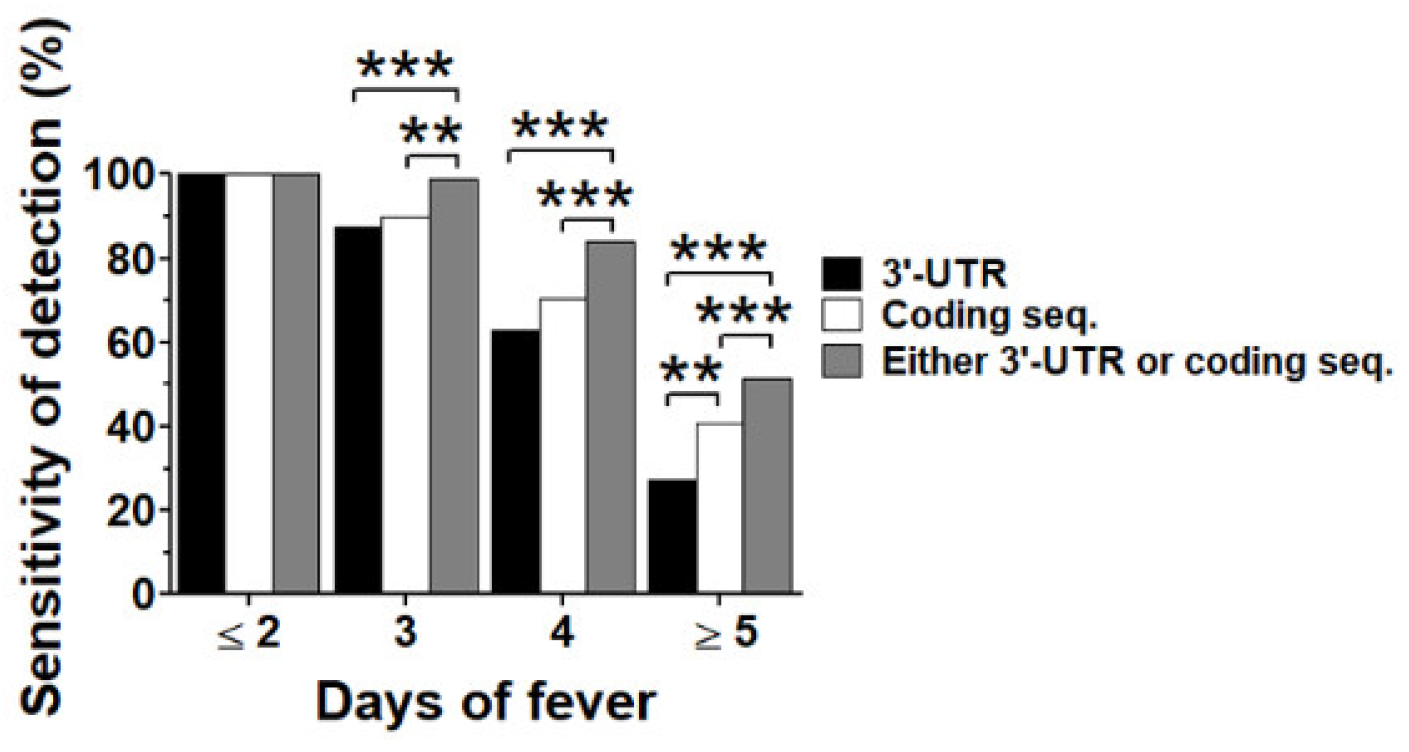
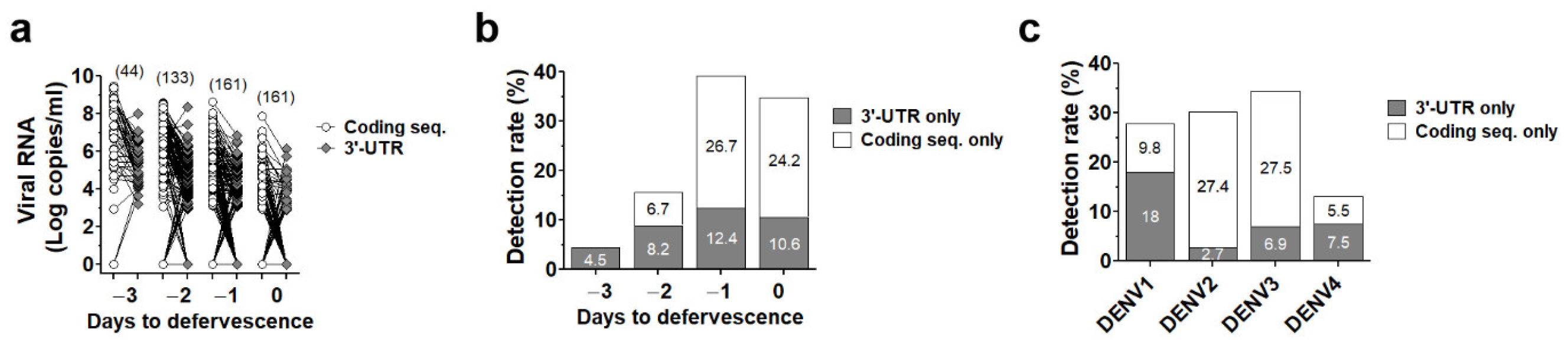
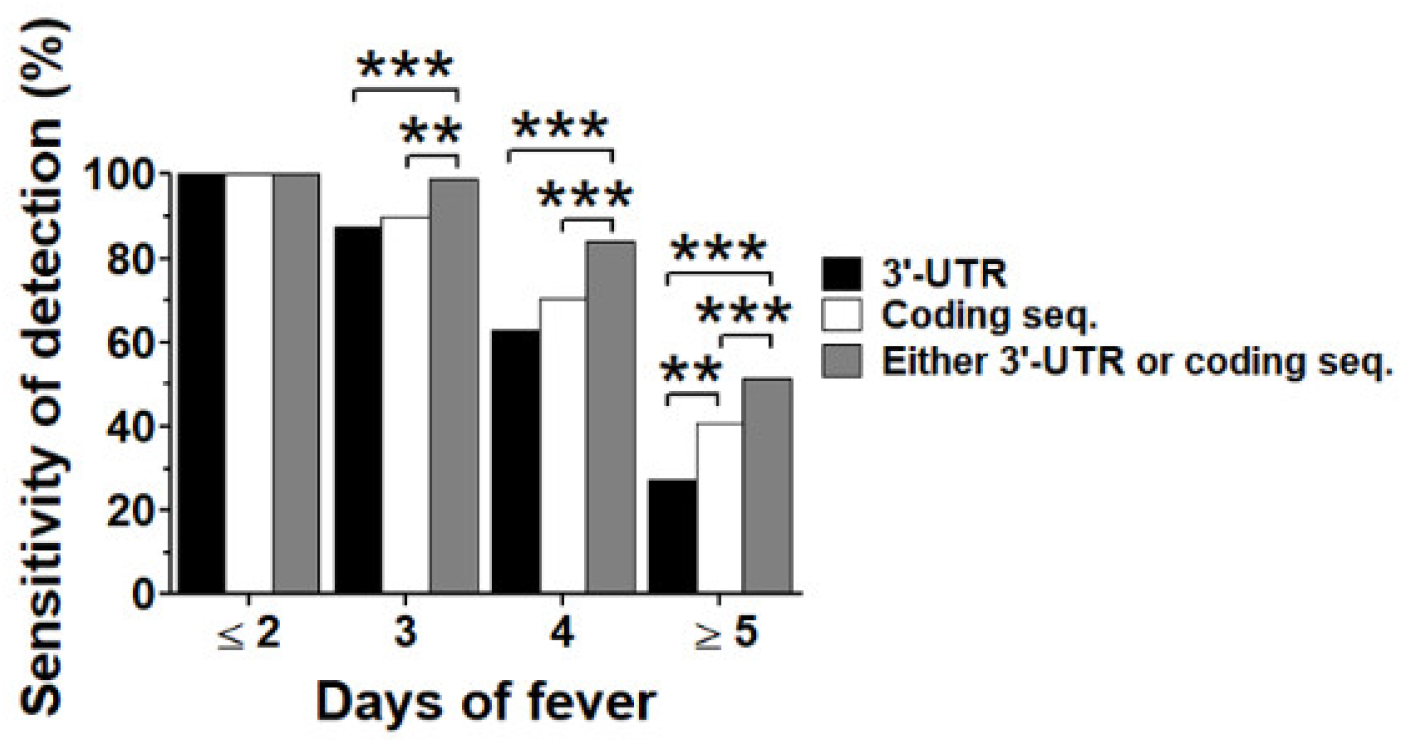
3.3. Dual Region Detection Enhanced Sensitivity of qRT-PCR Assay
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Brady, O.J.; Golding, N.; Kraemer, M.U.G.; Wint, G.R.W.; Ray, S.E.; Pigott, D.M.; Shearer, F.M.; Johnson, K.; Earl, L.; et al. The current and future global distribution and population at risk of dengue. Nat. Microbiol. 2019, 4, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Dengue Haemorrhagic Fever: Diagnosis, Treatment, Prevention and Control; World Health Organization: Geneva, Switzerland, 1997.
- Halstead, S.B.; Russell, P.K. Protective and immunological behavior of chimeric yellow fever dengue vaccine. Vaccine 2016, 34, 1643–1647. [Google Scholar] [CrossRef]
- Hunsperger, E.A.; Yoksan, S.; Buchy, P.; Nguyen, V.C.; Sekaran, S.D.; Enria, D.A.; Pelegrino, J.L.; Vazquez, S.; Artsob, H.; Drebot, M.; et al. Evaluation of commercially available anti-dengue virus immunoglobulin M tests. Emerg. Infect. Dis. 2009, 15, 436–440. [Google Scholar] [CrossRef]
- Peeling, R.W.; Artsob, H.; Pelegrino, J.L.; Buchy, P.; Cardosa, M.J.; Devi, S.; Enria, D.A.; Farrar, J.; Gubler, D.J.; Guzman, M.G.; et al. Evaluation of diagnostic tests: Dengue. Nat. Rev. Microbiol. 2010, 8, S30–S37. [Google Scholar] [CrossRef]
- Dussart, P.; Petit, L.; Labeau, B.; Bremand, L.; Leduc, A.; Moua, D.; Matheus, S.; Baril, L. Evaluation of two new commercial tests for the diagnosis of acute dengue virus infection using NS1 antigen detection in human serum. PLoS Negl. Trop. Dis. 2008, 2, e280. [Google Scholar] [CrossRef]
- Namekar, M.; Ellis, E.M.; O’Connell, M.; Elm, J.; Gurary, A.; Park, S.Y.; Imrie, A.; Nerurkar, V.R. Evaluation of a new commercially available immunoglobulin M capture enzyme-linked immunosorbent assay for diagnosis of dengue virus infection. J. Clin. Microbiol. 2013, 51, 3102–3106. [Google Scholar] [CrossRef]
- Anderson, N.W.; Jespersen, D.J.; Rollins, L.; Seaton, B.; Prince, H.E.; Theel, E.S. Detection of the dengue virus NS1 antigen using an enzyme immunoassay. Diagn. Microbiol. Infect. Dis. 2014, 79, 194–197. [Google Scholar] [CrossRef]
- Pal, S.; Dauner, A.L.; Mitra, I.; Forshey, B.M.; Garcia, P.; Morrison, A.C.; Halsey, E.S.; Kochel, T.J.; Wu, S.J. Evaluation of dengue NS1 antigen rapid tests and ELISA kits using clinical samples. PLoS ONE 2014, 9, e113411. [Google Scholar] [CrossRef] [PubMed]
- Vivek, R.; Ahamed, S.F.; Kotabagi, S.; Chandele, A.; Khanna, I.; Khanna, N.; Nayak, K.; Dias, M.; Kaja, M.K.; Shet, A. Evaluation of a pan-serotype point-of-care rapid diagnostic assay for accurate detection of acute dengue infection. Diagn. Microbiol. Infect. Dis. 2017, 87, 229–234. [Google Scholar] [CrossRef]
- Kabir, M.A.; Zilouchian, H.; Younas, M.A.; Asghar, W. Dengue Detection: Advances in Diagnostic Tools from Conventional Technology to Point of Care. Biosensors 2021, 11, 206. [Google Scholar] [CrossRef]
- Lau, Y.L.; Lai, M.Y.; Teoh, B.T.; Abd-Jamil, J.; Johari, J.; Sam, S.S.; Tan, K.K.; AbuBakar, S. Colorimetric Detection of Dengue by Single Tube Reverse-Transcription-Loop-Mediated Isothermal Amplification. PLoS ONE 2015, 10, e0138694. [Google Scholar] [CrossRef]
- Neeraja, M.; Lakshmi, V.; Lavanya, V.; Priyanka, E.N.; Parida, M.M.; Dash, P.K.; Sharma, S.; Rao, P.V.; Reddy, G. Rapid detection and differentiation of dengue virus serotypes by NS1 specific reverse transcription loop-mediated isothermal amplification (RT-LAMP) assay in patients presenting to a tertiary care hospital in Hyderabad, India. J. Virol. Methods 2015, 211, 22–31. [Google Scholar] [CrossRef]
- Gao, M.; Waggoner, J.J.; Hecht, S.M.; Chen, S. Selective Detection of Dengue Virus Serotypes Using Tandem Toehold-Mediated Displacement Reactions. ACS Infect. Dis. 2019, 5, 1907–1914. [Google Scholar] [CrossRef]
- Gao, M.; Daniel, D.; Zou, H.; Jiang, S.; Lin, S.; Huang, C.; Hecht, S.M.; Chen, S. Rapid detection of a dengue virus RNA sequence with single molecule sensitivity using tandem toehold-mediated displacement reactions. Chem. Commun. 2018, 54, 968–971. [Google Scholar] [CrossRef]
- Shu, P.Y.; Chang, S.F.; Kuo, Y.C.; Yueh, Y.Y.; Chien, L.J.; Sue, C.L.; Lin, T.H.; Huang, J.H. Development of group- and serotype-specific one-step SYBR green I-based real-time reverse transcription-PCR assay for dengue virus. J. Clin. Microbiol. 2003, 41, 2408–2416. [Google Scholar] [CrossRef]
- Johnson, B.W.; Russell, B.J.; Lanciotti, R.S. Serotype-specific detection of dengue viruses in a fourplex real-time reverse transcriptase PCR assay. J. Clin. Microbiol. 2005, 43, 4977–4983. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Thay, C.H.; Tin, T.C.; Devi, S. Rapid detection, serotyping and quantitation of dengue viruses by TaqMan real-time one-step RT-PCR. J. Virol. Methods 2006, 138, 123–130. [Google Scholar] [CrossRef]
- Hue, K.D.; Tuan, T.V.; Thi, H.T.; Bich, C.T.; Anh, H.H.; Wills, B.A.; Simmons, C.P. Validation of an internally controlled one-step real-time multiplex RT-PCR assay for the detection and quantitation of dengue virus RNA in plasma. J. Virol. Methods 2011, 177, 168–173. [Google Scholar] [CrossRef]
- Srikiatkhachorn, A.; Wichit, S.; Gibbons, R.V.; Green, S.; Libraty, D.H.; Endy, T.P.; Ennis, F.A.; Kalayanarooj, S.; Rothman, A.L. Dengue viral RNA levels in peripheral blood mononuclear cells are associated with disease severity and preexisting dengue immune status. PLoS ONE 2012, 7, e51335. [Google Scholar] [CrossRef]
- Waggoner, J.J.; Abeynayake, J.; Sahoo, M.K.; Gresh, L.; Tellez, Y.; Gonzalez, K.; Ballesteros, G.; Pierro, A.M.; Gaibani, P.; Guo, F.P.; et al. Single-reaction, multiplex, real-time rt-PCR for the detection, quantitation, and serotyping of dengue viruses. PLoS Negl. Trop. Dis. 2013, 7, e2116. [Google Scholar] [CrossRef]
- Najioullah, F.; Viron, F.; Cesaire, R. Evaluation of four commercial real-time RT-PCR kits for the detection of dengue viruses in clinical samples. Virol. J. 2014, 11, 164. [Google Scholar] [CrossRef]
- Alm, E.; Lindegren, G.; Falk, K.I.; Lagerqvist, N. One-step real-time RT-PCR assays for serotyping dengue virus in clinical samples. BMC Infect. Dis. 2015, 15, 493. [Google Scholar] [CrossRef]
- Waggoner, J.J.; Ballesteros, G.; Gresh, L.; Mohamed-Hadley, A.; Tellez, Y.; Sahoo, M.K.; Abeynayake, J.; Balmaseda, A.; Harris, E.; Pinsky, B.A. Clinical evaluation of a single-reaction real-time RT-PCR for pan-dengue and chikungunya virus detection. J. Clin. Virol. 2016, 78, 57–61. [Google Scholar] [CrossRef]
- Yang, L.; Liang, W.; Jiang, L.; Li, W.; Cao, W.; Wilson, Z.A.; Zhang, D. A novel universal real-time PCR system using the attached universal duplex probes for quantitative analysis of nucleic acids. BMC Mol. Biol. 2008, 9, 54. [Google Scholar] [CrossRef]
- Brownie, J.; Shawcross, S.; Theaker, J.; Whitcombe, D.; Ferrie, R.; Newton, C.; Little, S. The elimination of primer-dimer accumulation in PCR. Nucleic Acids Res. 1997, 25, 3235–3241. [Google Scholar] [CrossRef]
- Polz, M.F.; Cavanaugh, C.M. Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 1998, 64, 3724–3730. [Google Scholar] [CrossRef]
- Prada-Arismendy, J.; Rincon, V.; Castellanos, J.E. Comparative evaluation of permissiveness to dengue virus serotype 2 infection in primary rodent macrophages. J. Trop. Med. 2012, 2012, 950303. [Google Scholar] [CrossRef]
- Yenchitsomanus, P.T.; Sricharoen, P.; Jaruthasana, I.; Pattanakitsakul, S.N.; Nitayaphan, S.; Mongkolsapaya, J.; Malasit, P. Rapid detection and identification of dengue viruses by polymerase chain reaction (PCR). S. Asian J. Trop. Med. Public Health 1996, 27, 228–236. [Google Scholar]
- Punyadee, N.; Mairiang, D.; Thiemmeca, S.; Komoltri, C.; Pan-Ngum, W.; Chomanee, N.; Charngkaew, K.; Tangthawornchaikul, N.; Limpitikul, W.; Vasanawathana, S.; et al. Microparticles provide a novel biomarker to predict severe clinical outcomes of dengue virus infection. J. Virol. 2015, 89, 1587–1607. [Google Scholar] [CrossRef]
- Lin, C.W.; Wu, S.C. A functional epitope determinant on domain III of the Japanese encephalitis virus envelope protein interacted with neutralizing-antibody combining sites. J. Virol. 2003, 77, 2600–2606. [Google Scholar] [CrossRef]
- Faye, O.; Faye, O.; Dupressoir, A.; Weidmann, M.; Ndiaye, M.; Alpha Sall, A. One-step RT-PCR for detection of Zika virus. J. Clin. Virol. 2008, 43, 96–101. [Google Scholar] [CrossRef]
- Innis, B.L.; Nisalak, A.; Nimmannitya, S.; Kusalerdchariya, S.; Chongswasdi, V.; Suntayakorn, S.; Puttisri, P.; Hoke, C.H. An enzyme-linked immunosorbent assay to characterize dengue infections where dengue and Japanese encephalitis co-circulate. Am. J. Trop. Med. Hyg. 1989, 40, 418–427. [Google Scholar] [CrossRef]
- Puttikhunt, C.; Prommool, T.; Nathaporn, U.; Ong-ajchaowlerd, P.; Yoosook, K.; Tawilert, C.; Duangchinda, T.; Jairangsri, A.; Tangthawornchaikul, N.; Malasit, P.; et al. The development of a novel serotyping-NS1-ELISA to identify serotypes of dengue virus. J. Clin. Virol. 2011, 50, 314–319. [Google Scholar] [CrossRef]
- Hall, T.A. Bioedit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 4 May 2018).
- Avirutnan, P.; Punyadee, N.; Noisakran, S.; Komoltri, C.; Thiemmeca, S.; Auethavornanan, K.; Jairungsri, A.; Kanlaya, R.; Tangthawornchaikul, N.; Puttikhunt, C.; et al. Vascular leakage in severe dengue virus infections: A potential role for the nonstructural viral protein NS1 and complement. J. Infect. Dis. 2006, 193, 1078–1088. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]
- Brooks, E.M.; Sheflin, L.G.; Spaulding, S.W. Secondary structure in the 3’ UTR of EGF and the choice of reverse transcriptases affect the detection of message diversity by RT-PCR. Biotechniques 1995, 19, 806–812. [Google Scholar]
- Aaskov, J.; Buzacott, K.; Thu, H.M.; Lowry, K.; Holmes, E.C. Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science 2006, 311, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lott, W.B.; Lowry, K.; Jones, A.; Thu, H.M.; Aaskov, J. Defective interfering viral particles in acute dengue infections. PLoS ONE 2011, 6, e19447. [Google Scholar] [CrossRef]
- Liu, R.; Yue, L.; Li, X.; Yu, X.; Zhao, H.; Jiang, Z.; Qin, E.; Qin, C. Identification and characterization of small sub-genomic RNAs in dengue 1-4 virus-infected cell cultures and tissues. Biochem. Biophys. Res. Commun. 2010, 391, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Aaskov, J. Sub-genomic RNA of defective interfering (D.I.) dengue viral particles is replicated in the same manner as full length genomes. Virology 2014, 468–470, 248–255. [Google Scholar] [CrossRef]
- Pesko, K.N.; Fitzpatrick, K.A.; Ryan, E.M.; Shi, P.Y.; Zhang, B.; Lennon, N.J.; Newman, R.M.; Henn, M.R.; Ebel, G.D. Internally deleted WNV genomes isolated from exotic birds in New Mexico: Function in cells, mosquitoes, and mice. Virology 2012, 427, 10–17. [Google Scholar] [CrossRef]
- Sun, Y.; Jain, D.; Koziol-White, C.J.; Genoyer, E.; Gilbert, M.; Tapia, K.; Panettieri, R.A., Jr.; Hodinka, R.L.; Lopez, C.B. Immunostimulatory Defective Viral Genomes from Respiratory Syncytial Virus Promote a Strong Innate Antiviral Response during Infection in Mice and Humans. PLoS Pathog. 2015, 11, e1005122. [Google Scholar] [CrossRef]
- Vasilijevic, J.; Zamarreno, N.; Oliveros, J.C.; Rodriguez-Frandsen, A.; Gomez, G.; Rodriguez, G.; Perez-Ruiz, M.; Rey, S.; Barba, I.; Pozo, F.; et al. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog. 2017, 13, e1006650. [Google Scholar] [CrossRef]
- Li, Q.; Tong, Y.; Xu, Y.; Niu, J.; Zhong, J. Genetic Analysis of Serum-Derived Defective Hepatitis C Virus Genomes Revealed Novel Viral cis Elements for Virus Replication and Assembly. J. Virol. 2018, 92, e02182-17. [Google Scholar] [CrossRef]
- Simon, O.; Williams, T.; Caballero, P.; Lopez-Ferber, M. Dynamics of deletion genotypes in an experimental insect virus population. Proc. Biol. Sci. 2006, 273, 783–790. [Google Scholar] [CrossRef]
- Tapia, K.; Kim, W.K.; Sun, Y.; Mercado-Lopez, X.; Dunay, E.; Wise, M.; Adu, M.; Lopez, C.B. Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog. 2013, 9, e1003703. [Google Scholar] [CrossRef]
- Parameswaran, P.; Wang, C.; Trivedi, S.B.; Eswarappa, M.; Montoya, M.; Balmaseda, A.; Harris, E. Intrahost Selection Pressures Drive Rapid Dengue Virus Microevolution in Acute Human Infections. Cell Host Microbe 2017, 22, 400–410.e5. [Google Scholar] [CrossRef] [PubMed]
- Poirier, E.Z.; Goic, B.; Tome-Poderti, L.; Frangeul, L.; Boussier, J.; Gausson, V.; Blanc, H.; Vallet, T.; Loyd, H.; Levi, L.I.; et al. Dicer-2-Dependent Generation of Viral DNA from Defective Genomes of RNA Viruses Modulates Antiviral Immunity in Insects. Cell Host Microbe 2018, 23, 353–365.e8. [Google Scholar] [CrossRef] [PubMed]
- Linder, A.; Bothe, V.; Linder, N.; Schwarzlmueller, P.; Dahlstrom, F.; Bartenhagen, C.; Dugas, M.; Pandey, D.; Thorn-Seshold, J.; Boehmer, D.F.R.; et al. Defective Interfering Genomes and the Full-Length Viral Genome Trigger RIG-I After Infection With Vesicular Stomatitis Virus in a Replication Dependent Manner. Front. Immunol. 2021, 12, 595390. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Patients (n = 161) |
|---|---|
| Gender (male:female) | 95:66 |
| Age [mean ± SD (min–max)] | 10.6 ± 2.6 (5–15) |
| First date of collection [median (min–max)] | |
| Day of illness | 3 (0–7) |
| Day to defervescence * | −2 (−3–−1) |
| Last date of collection [median (min–max)] | |
| Day of illness | 6 (1–9) |
| Day to defervescence | 0 (0) |
| Severity (DF:DHF1:DHF2:DHF3) # | 80:22:53:6 |
| DENV infection (primary:secondary) | 0:161 |
| DENV1 (collected from year 2008 to 2013) | 39 |
| DENV2 (collected from year 2006 to 2012) | 39 |
| DENV3 (collected from year 2010 to 2013) | 39 |
| DENV4 (collected from year 2006 to 2013) | 44 |
| No. | Primers/Probes | Nucleotide Sequences (5′–3′) | Nucleotide No. (Region) | |
|---|---|---|---|---|
| 1 |  | F primer | GGTTAGAGGAGACCCCTCCC | 10424–10443 (all DENV-3′-UTR) |
| R primer | GGCGY # TCTGTGCCTGGA | 10596–10612 (all DENV-3′-UTR) | ||
| Probe | 6-FAM-CAGGATCTCTGGTCTY # TCCCAGCGT–BHQ | 10553–10577 (all DENV-3′-UTR) | ||
| 2 | | F primer | CAAAAGGAAGTCGTGCAATA | 8974–8993 (DENV1-NS5) |
| R primer | CTGAGTGAATTCTCTCTR $ CTGAACC | 9061–9085 (DENV1-NS5) | ||
| Probe | 6-FAM-CATGTGGTTGGGAGCACGC–BHQ | 8999–9017 (DENV1-NS5) | ||
| 3 | | F primer | CAGGTTATGGCACY # GTCACR $ AT | 1463–1484 (DENV2-E) |
| R primer | CCATCTGCAGCAACACCATCTC | 1519–1540 (DENV2-E) | ||
| Probe | 6-FAM-CTCY # CCGAGAACAGGCCTCGACTTCAA–BHQ | 1491–1517 (DENV2-E) | ||
| 4 | | F primer | GGACTGGACACACGCACY # CA | 740–759 (DENV3-M) |
| R primer | CATGTCTCTACCTTCTCGACTTGTCT | 788–813 (DENV3-M) | ||
| Probe | 6-FAM-ACCTGGATGTCGGCY # GAAGGAGCTTG–BHQ | 761–786 (DENV3-M) | ||
| 5 | | F primer | TTGTY # CTAATGATGCTN & GTCG | 896–916 (DENV4-M/E) |
| R primer | TCCACCTGAGACTCCTTCY # A | 965–984 (DENV4-M/E) | ||
| Probe | 6-FAM-TY # CCY # ACTCCTACGCATCGCATTCCG–BHQ | 927–952 (DENV4-M/E) | ||
| Serotypes | Copies/Reaction | Positive/Total | Detection Rate (%) | Mean ± SD of Ct Values | % CV of Ct Values |
|---|---|---|---|---|---|
| DENV1 | 100 | 8/8 | 100 | 33.2 ± 0.5 | 1.6 |
| 10 * | 8/8 | 100 * | 36.3 ± 0.8 | 2.3 # | |
| 5 | 0/8 | 0 | UD 1 | - | |
| DENV2 | 100 | 8/8 | 100 | 31.6 ± 0.3 | 1.0 |
| 10 * | 8/8 | 100 * | 35.2 ± 0.5 | 1.5 # | |
| 5 | 0/8 | 0 | UD | - | |
| DENV3 | 100 | 8/8 | 100 | 32.8 ± 1.2 | 3.5 |
| 10 * | 8/8 | 100 * | 36.9 ± 1.0 | 2.7 # | |
| 5 | 0/8 | 0 | UD | - | |
| DENV4 | 100 | 8/8 | 100 | 33.1 ± 1.7 | 5.3 |
| 10 * | 8/8 | 100 * | 35.8 ± 1.6 | 4.4 # | |
| 5 | 0/8 | 0 | UD | - |
| DENV Serotypes | Total Patient Number | DENV Positive Number | Sensitivity of Detection (%) |
|---|---|---|---|
| All serotypes | 161 | 156 | 96.9 |
| DENV1 | 39 | 39 | 100.0 |
| DENV2 | 39 | 39 | 100.0 |
| DENV3 | 39 | 39 | 100.0 |
| DENV4 | 44 | 39 | 88.6 |
| Day of Fever | Total Specimens | DENV Positive Specimens | Sensitivity of Detection (%) |
|---|---|---|---|
| ≤2 | 79 | 79 | 100 |
| 3 | 112 | 98 | 87.5 |
| 4 | 145 | 91 | 62.8 |
| ≥5 | 192 | 52 | 27.1 |
| Serotypes | Copies/Reaction | Positive/Total | Positivity Rate (%) | Mean ± SD of Ct Values | % CV of Ct Values |
|---|---|---|---|---|---|
| DENV1 | 100 | 8/8 | 100 | 29.3 ± 1.6 | 5.4 |
| 10 | 8/8 | 100 * | 32.6 ± 1.5 | 4.7 # | |
| 5 | 5/8 | 63 | NA 1 | - | |
| DENV2 | 100 | 8/8 | 100 | 31.6 ± 0.3 | 2.8 |
| 10 | 8/8 | 100 * | 35.2 ± 0.5 | 3.1 # | |
| 5 | 1/8 | 13 | NA | - | |
| DENV3 | 100 | 8/8 | 100 | 32.8 ± 1.2 | 2.4 |
| 10 | 8/8 | 100 * | 36.9 ± 1.0 | 2.4 # | |
| 5 | 7/8 | 88 | NA | - | |
| DENV4 | 100 | 8/8 | 100 | 33.1 ± 1.7 | 4.2 |
| 10 | 8/8 | 100 * | 35.8 ± 1.6 | 2.0 # | |
| 5 | 7/8 | 88 | NA | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Songjaeng, A.; Thiemmeca, S.; Mairiang, D.; Punyadee, N.; Kongmanas, K.; Hansuealueang, P.; Tangthawornchaikul, N.; Duangchinda, T.; Mongkolsapaya, J.; Sriruksa, K.; et al. Development of a Singleplex Real-Time Reverse Transcriptase PCR Assay for Pan-Dengue Virus Detection and Quantification. Viruses 2022, 14, 1271. https://doi.org/10.3390/v14061271
Songjaeng A, Thiemmeca S, Mairiang D, Punyadee N, Kongmanas K, Hansuealueang P, Tangthawornchaikul N, Duangchinda T, Mongkolsapaya J, Sriruksa K, et al. Development of a Singleplex Real-Time Reverse Transcriptase PCR Assay for Pan-Dengue Virus Detection and Quantification. Viruses. 2022; 14(6):1271. https://doi.org/10.3390/v14061271
Chicago/Turabian StyleSongjaeng, Adisak, Somchai Thiemmeca, Dumrong Mairiang, Nuntaya Punyadee, Kessiri Kongmanas, Prachya Hansuealueang, Nattaya Tangthawornchaikul, Thaneeya Duangchinda, Juthathip Mongkolsapaya, Kanokwan Sriruksa, and et al. 2022. "Development of a Singleplex Real-Time Reverse Transcriptase PCR Assay for Pan-Dengue Virus Detection and Quantification" Viruses 14, no. 6: 1271. https://doi.org/10.3390/v14061271
APA StyleSongjaeng, A., Thiemmeca, S., Mairiang, D., Punyadee, N., Kongmanas, K., Hansuealueang, P., Tangthawornchaikul, N., Duangchinda, T., Mongkolsapaya, J., Sriruksa, K., Limpitikul, W., Malasit, P., & Avirutnan, P. (2022). Development of a Singleplex Real-Time Reverse Transcriptase PCR Assay for Pan-Dengue Virus Detection and Quantification. Viruses, 14(6), 1271. https://doi.org/10.3390/v14061271