
Complete Genome Sequencing of Tick-Borne Encephalitis Virus Directly from Clinical Samples: Comparison of Shotgun Metagenomic and Targeted Amplicon-Based Sequencing

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. TBE Infection Case Definition
2.3. Ethics Considerations
2.4. Available TBEV Genome Sequences
2.5. Viral Load
2.6. Design of Oligonucleotide Primer Pairs for Amplicon Amplification of TBEV Genome
2.7. PCR Amplification and Optimization of Primer Pools
2.8. Sample Preparation for Shotgun Metagenomic Sequencing
2.9. Sequencing Using Illumina
2.10. Sequencing with Oxford Nanopore Technologies
2.11. Bioinformatic Analysis
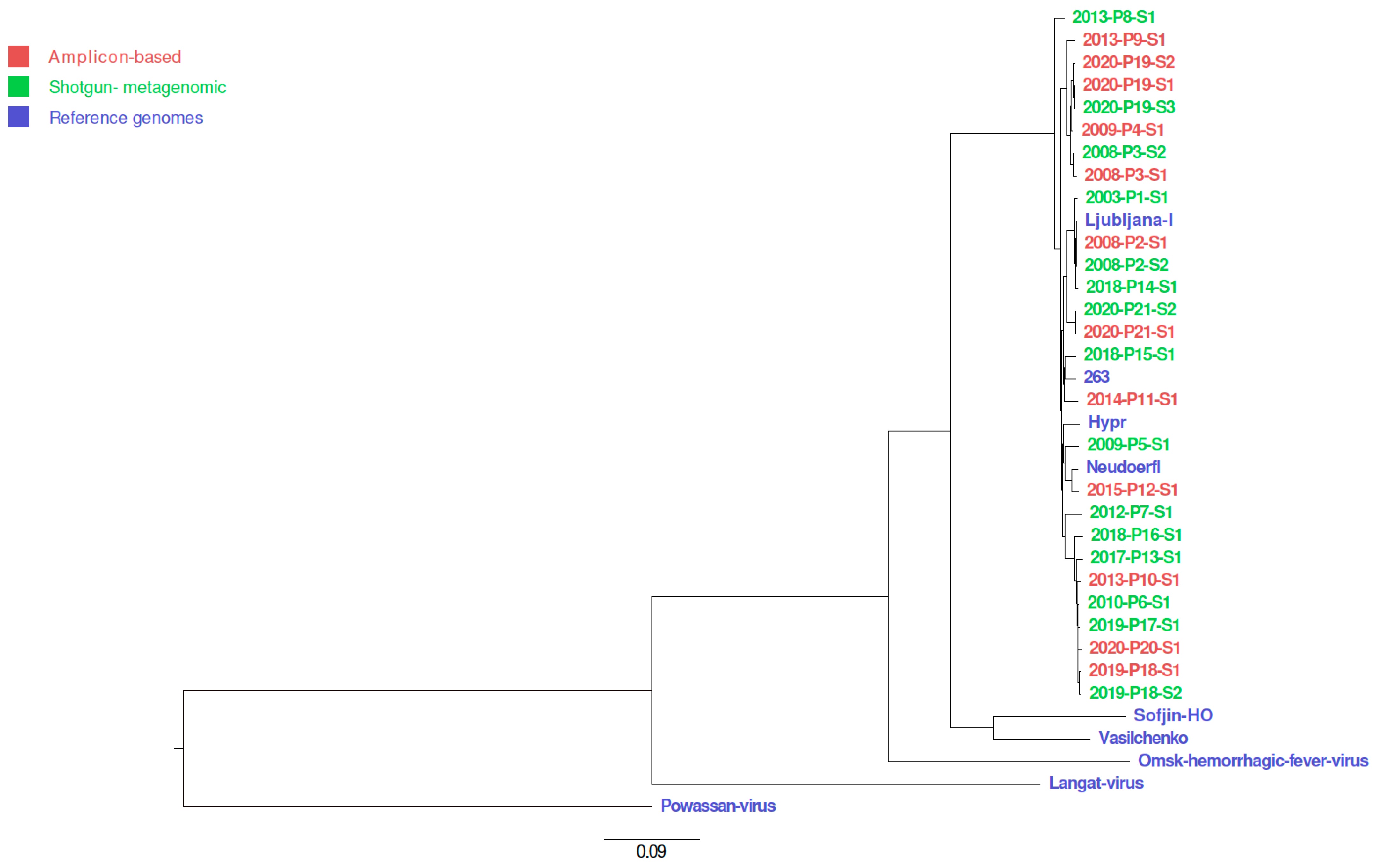
2.12. Phylogenetic Tree
3. Results
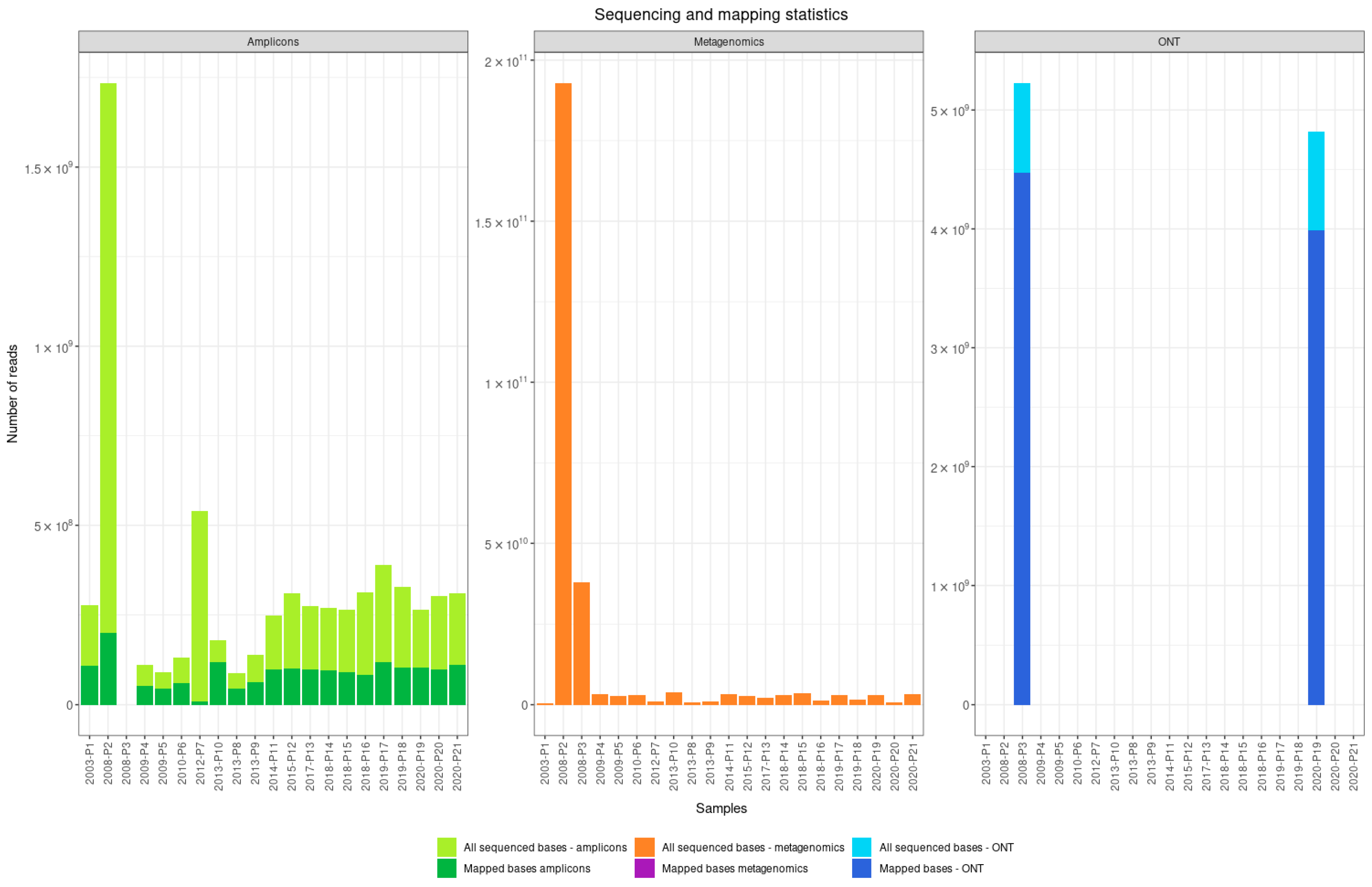
3.1. Comparison of Shotgun Metagenomic Sequencing and the Amplicon-Based Approach
3.2. Comparison of Consensus Sequences of Nearly Complete Genomes Generated with Two Approaches
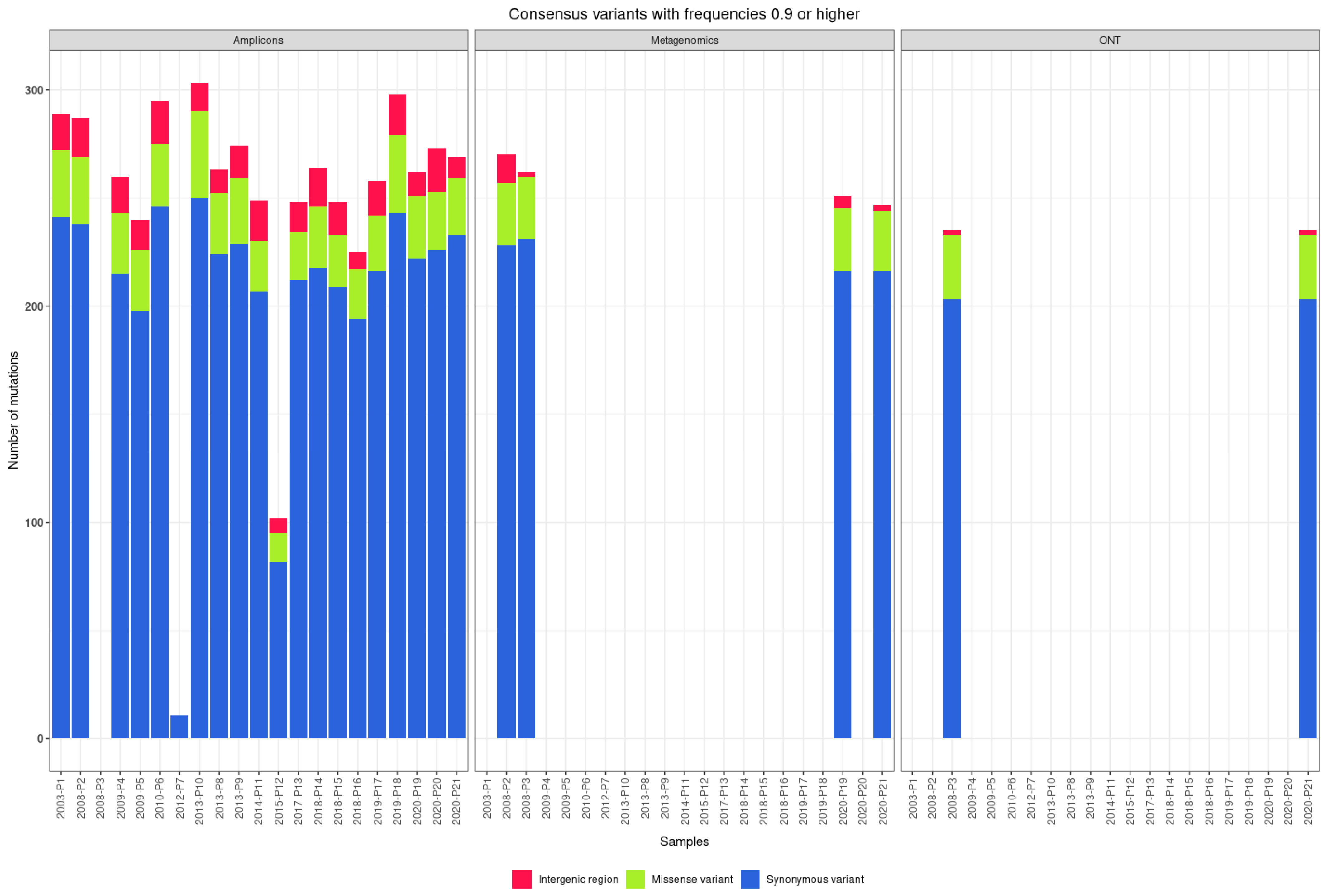
3.3. Consensus Variants
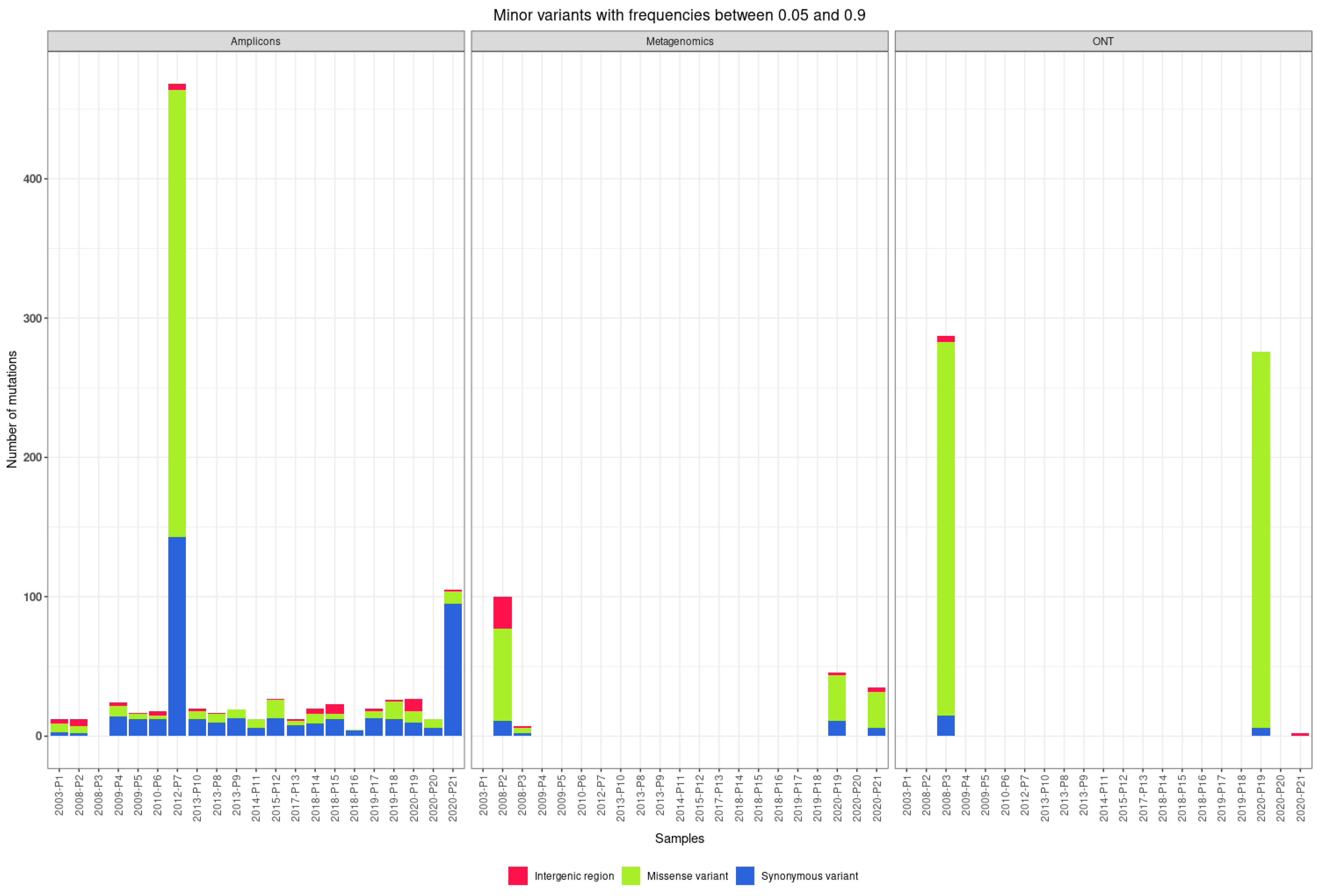
3.4. Quasispecies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ecker, M.; Allison, S.L.; Meixner, T.; Heinz, F.X. Sequence Analysis and Genetic Classification of Tick-Borne Encephalitis Viruses from Europe and Asia. J. Gen. Virol. 1999, 80, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Valeryevna, K.I.; Vasilievna, D.T.; Evgenyevich, T.S.; Konstantinovna, D.E.; Vasilievna, L.O.; Mihailovna, V.M.; Stanislavovna, K.L.; Pavlovich, D.Y.; Igorevich, P.A.; Vladimirovna, S.O.; et al. Characteristics of the Baikal Subtype of Tick-Borne Encephalitis Virus Circulating in Eastern Siberia. Acta Biomed. Sci. 2018, 3, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Shang, G.; Lu, S.; Yang, J.; Xu, J. A New Subtype of Eastern Tick-Borne Encephalitis Virus Discovered in Qinghai-Tibet Plateau, China Article. Emerg. Microbes Infect. 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Lindquist, L.; Vapalahti, O. Tick-Borne Encephalitis. Lancet 2008, 371, 1861–1871. [Google Scholar] [CrossRef]
- Gritsun, T.S.; Lashkevich, V.A.; Gould, E.A. Tick-Borne Encephalitis. Antiviral Res. 2003, 57, 129–146. [Google Scholar] [CrossRef]
- Ličková, M.; Havlíková, S.F.; Sláviková, M.; Klempa, B. Alimentary Infections by Tick-Borne Encephalitis Virus. Viruses 2021, 14, 56. [Google Scholar] [CrossRef]
- Lipowski, D.; Popiel, M.; Perlejewski, K.; Nakamura, S.; Bukowska-Ośko, I.; Rzadkiewicz, E.; Dzieciątkowski, T.; Milecka, A.; Wenski, W.; Ciszek, M.; et al. A Cluster of Fatal Tick-Borne Encephalitis Virus Infection in Organ Transplant Setting. J. Infect. Dis. 2017, 215, 896–901. [Google Scholar] [CrossRef] [Green Version]
- Avšič-Županc, T.; Poljak, M.; Matičič, M.; Radšel-Medvešček, A.; LeDuc, J.W.; Stiasny, K.; Kunz, C.; Heinz, F.X. Laboratory Acquired Tick-Borne Meningoencephalitis: Characterisation of Virus Strains. Clin. Diagn. Virol. 1995, 4, 51–59. [Google Scholar] [CrossRef]
- Hoogstraal, H. Ticks in Relation to Human Diseases Caused by Viruses. Annu. Rev. Entomol. 1966, 11, 261–308. [Google Scholar] [CrossRef]
- Rubel, F.; Brugger, K.; Walter, M.; Vogelgesang, J.R.; Didyk, Y.M.; Fu, S.; Kahl, O. Geographical Distribution, Climate Adaptation and Vector Competence of the Eurasian Hard Tick Haemaphysalis Concinna. Ticks Tick. Borne. Dis. 2018, 9, 1080–1089. [Google Scholar] [CrossRef]
- Pulkkinen, L.I.A.; Butcher, S.J.; Anastasina, M. Tick-Borne Encephalitis Virus: A Structural View. Viruses 2018, 10, 350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogovič, P.; Lotrič-Furlan, S.; Avšič-Županc, T.; Lusa, L.; Strle, F. Factors Associated with Severity of Tick-Borne Encephalitis: A Prospective Observational Study. Travel Med. Infect. Dis. 2018, 26, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Bogovič, P.; Lotrič-Furlan, S.; Avšič-županc, T.; Korva, M.; Kastrin, A.; Lusa, L.; Strle, K.; Strle, F. Comparison of Clinical, Laboratory and Immune Characteristics of the Monophasic and Biphasic Course of Tick-Borne Encephalitis. Microorganisms 2021, 9, 796. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R. Tick-Borne Encephalitis. Infect. Dis. Clin. North Am. 2008, 22, 561–575. [Google Scholar] [CrossRef]
- Ternovoi, V.A.; Kurzhukov, G.P.; Sokolov, Y.V.; Ivanov, G.Y.; Ivanisenko, V.A.; Loktev, A.V.; Ryder, R.W.; Netesov, S.V.; Loktev, V.B. Tick-Borne Encephalitis with Hemorrhagic Syndrome, Novosibirsk Region, Russia, 1999—Volume 9, Number 6—June 2003—Emerging Infectious Diseases Journal—CDC. Emerg. Infect. Dis. 2003, 9, 743–746. [Google Scholar] [CrossRef]
- National Institute of Public Health NIJZ Data Portal. Available online: https://podatki.nijz.si/pxweb/sl/NIJZ podatkovniportal/?px_language=sl&px_db=NIJZpodatkovni portal&rxid=6cfa1ca6-229f-40d4-8e42-fd07c23a250a (accessed on 9 January 2020).
- Fajs, L.; Durmiši, E.; Knap, N.; Strle, F.; Avšič-Županc, T. Phylogeographic Characterization of Tick-Borne Encephalitis Virus from Patients, Rodents and Ticks in Slovenia. PLoS ONE 2012, 7, e48420. [Google Scholar] [CrossRef]
- Saksida, A.; Jakopin, N.; Jelovšek, M.; Knap, N.; Fajs, L.; Lusa, L.; Lotrič-Furlan, S.; Bogovič, P.; Arnež, M.; Strle, F.; et al. Virus RNA Load in Patients with Tick-Borne Encephalitis, Slovenia. Emerg. Infect. Dis. 2018, 24, 1315–1323. [Google Scholar] [CrossRef] [Green Version]
- Bogovič, P.; Kastrin, A.; Lotrič-Furlan, S.; Ogrinc, K.; Županc, T.A.; Korva, M.; Knap, N.; Strle, F. Clinical and Laboratory Characteristics and Outcome of Illness Caused by Tick-Borne Encephalitis Virus without Central Nervous System Involvement—Volume 28, Number 2—February 2022—Emerging Infectious Diseases Journal—CDC. Emerg. Infect. Dis. 2022, 28, 291–301. [Google Scholar] [CrossRef]
- Giani, A.M.; Gallo, G.R.; Gianfranceschi, L.; Formenti, G. Long Walk to Genomics: History and Current Approaches to Genome Sequencing and Assembly. Comput. Struct. Biotechnol. J. 2020, 18, 9–19. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Isolating, Cloning, and Sequencing DNA; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Helmová, R.; Hönig, V.; Tykalová, H.; Palus, M.; Bell-Sakyi, L.; Grubhoffer, L. Tick-Borne Encephalitis Virus Adaptation in Different Host Environments and Existence of Quasispecies. Viruses 2020, 12, 902. [Google Scholar] [CrossRef]
- Boers, S.A.; Jansen, R.; Hays, J.P. Understanding and Overcoming the Pitfalls and Biases of Next-Generation Sequencing (NGS) Methods for Use in the Routine Clinical Microbiological Diagnostic Laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerome, H.; Taylor, C.; Sreenu, V.B.; Klymenko, T.; Filipe, A.D.S.; Jackson, C.; Davis, C.; Ashraf, S.; Wilson-Davies, E.; Jesudason, N.; et al. Metagenomic Next-Generation Sequencing Aids the Diagnosis of Viral Infections in Febrile Returning Travellers. J. Infect. 2019, 79, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.R.; Sundararaju, S.; Tang, P.; Tsui, K.M.; Lopez, A.P.; Janahi, M.; Tan, R.; Tilley, P. A Metagenomics-Based Diagnostic Approach for Central Nervous System Infections in Hospital Acute Care Setting. Sci. Rep. 2020, 10, 11194. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Semeraro, R.; Mingrino, A.; Giusti, B.; D’Aurizio, R. Nanopore Sequencing Data Analysis: State of the Art, Applications and Challenges. Brief. Bioinform. 2017, 19, 1256–1272. [Google Scholar] [CrossRef]
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Conlan, S.; Quan, P.-L.; Hui, J.; Marshall, J.; et al. A New Arenavirus in a Cluster of Fatal Transplant-Associated Diseases. N. Engl. J. Med. 2008, 358, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-Time, Portable Genome Sequencing for Ebola Surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR Method for MinION and Illumina Sequencing of Zika and Other Virus Genomes Directly from Clinical Samples. Nat. Protoc. 2017, 12, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Chaintoutis, S.C.; Papadopoulou, E.; Melidou, A.; Papa, A.; Dovas, C.I. A PCR-Based NGS Protocol for Whole Genome Sequencing of West Nile Virus Lineage 2 Directly from Biological Specimens. Mol. Cell. Probes 2019, 46, 101412. [Google Scholar] [CrossRef]
- Paulsen, K.M.; Lamsal, A.; Bastakoti, S.; Pettersson, J.H.O.; Pedersen, B.N.; Stiasny, K.; Haglund, M.; Smura, T.; Vapalahti, O.; Vikse, R.; et al. High-Throughput Sequencing of Two European Strains of Tick-Borne Encephalitis Virus (TBEV), Hochosterwitz and 1993/783. Ticks Tick. Borne. Dis. 2021, 12, 101557. [Google Scholar] [CrossRef]
- Worobey, M.; Watts, T.D.; McKay, R.A.; Suchard, M.A.; Granade, T.; Teuwen, D.E.; Koblin, B.A.; Heneine, W.; Lemey, P.; Jaffe, H.W. 1970s and ‘Patient 0’ HIV-1 Genomes Illuminate Early HIV/AIDS History in North America. Nature 2016, 539, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Schwaiger, M.; Cassinotti, P. Development of a Quantitative Real-Time RT-PCR Assay with Internal Control for the Laboratory Detection of Tick Borne Encephalitis Virus (TBEV) RNA. J. Clin. Virol. 2003, 27, 136–145. [Google Scholar] [CrossRef]
- BBDuk Guide—DOE Joint Genome Institute. Available online: https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk-guide/ (accessed on 15 November 2021).
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 16 April 2021).
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An Amplicon-Based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and IVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A. AliView: A Fast and Lightweight Alignment Viewer and Editor for Large Datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 25 May 2022).
- Petrackova, A.; Vasinek, M.; Sedlarikova, L.; Dyskova, T.; Schneiderova, P.; Novosad, T.; Papajik, T.; Kriegova, E. Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics. Front. Oncol. 2019, 9, 851. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, E.P.; Ternovoi, V.A.; Mikryukova, T.P.; Protopopova, E.V.; Gladysheva, A.V.; Shvalov, A.N.; Konovalova, S.N.; Chausov, E.V.; Loktev, V.B. Adaptation of Tick-Borne Encephalitis Virus from Human Brain to Different Cell Cultures Induces Multiple Genomic Substitutions. Arch. Virol. 2017, 162, 3151–3156. [Google Scholar] [CrossRef] [PubMed]
- Kuivanen, S.; Smura, T.; Rantanen, K.; Kämppi, L.; Kantonen, J.; Kero, M.; Jääskeläinen, A.; Jääskeläinen, A.J.; Sane, J.; Myllykangas, L.; et al. Fatal Tick-Borne Encephalitis Virus Infections Caused by Siberian and European Subtypes, Finland, 2015. Emerg. Infect. Dis. 2018, 24, 946–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulakova, N.V.; Andaev, E.I.; Belikov, S.I. Tick-Borne Encephalitis Virus in Eastern Siberia: Complete Genome Characteristics. Arch. Virol. 2012, 157, 2253–2255. [Google Scholar] [CrossRef]
- Egyed, L.; Biksi, I.; Varga, T.; Zöldi, V.; Dán, Á. Analysing the Genomes of Two Tick-Borne Encephalitis Viruses Isolated in Hungary in 1952 and 2019. Ticks Tick. Borne. Dis. 2021, 12, 101806. [Google Scholar] [CrossRef]
- Zmurko, J.; Neyts, J.; Dallmeier, K. Flaviviral NS4b, Chameleon and Jack-in-the-Box Roles in Viral Replication and Pathogenesis, and a Molecular Target for Antiviral Intervention. Rev. Med. Virol. 2015, 25, 205–223. [Google Scholar] [CrossRef] [Green Version]
- Gritsun, D.J.; Jones, I.M.; Gould, E.A.; Gritsun, T.S. Molecular Archaeology of Flaviviridae Untranslated Regions: Duplicated RNA Structures in the Replication Enhancer of Flaviviruses and Pestiviruses Emerged via Convergent Evolution. PLoS ONE 2014, 9, e92056. [Google Scholar] [CrossRef]
- Thurner, C.; Witwer, C.; Hofacker, I.L.; Stadler, P.F. Conserved RNA Secondary Structures in Flaviviridae Genomes. J. Gen. Virol. 2004, 85, 1113–1124. [Google Scholar] [CrossRef]
- Sakai, M.; Yoshii, K.; Sunden, Y.; Yokozawa, K.; Hirano, M.; Kariwa, H. Variable Region of the 3’ UTR Is a Critical Virulence Factor in the Far-Eastern Subtype of Tick-Borne Encephalitis Virus in a Mouse Model. J. Gen. Virol. 2014, 95, 823–835. [Google Scholar] [CrossRef]
- Asghar, N.; Lindblom, P.; Melik, W.; Lindqvist, R.; Haglund, M.; Forsberg, P.; Överby, A.K.; Andreassen, Å.; Lindgren, P.E.; Johansson, M. Tick-Borne Encephalitis Virus Sequenced Directly from Questing and Blood-Feeding Ticks Reveals Quasispecies Variance. PLoS ONE 2014, 9, e103264. [Google Scholar] [CrossRef]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies Diversity Determines Pathogenesis through Cooperative Interactions in a Viral Population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Perales, C. Viral Quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef] [Green Version]
- Krehenwinkel, H.; Wolf, M.; Lim, J.Y.; Rominger, A.J.; Simison, W.B.; Gillespie, R.G. Estimating and Mitigating Amplification Bias in Qualitative and Quantitative Arthropod Metabarcoding. Sci. Rep. 2017, 7, 17668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawluczyk, M.; Weiss, J.; Links, M.G.; Egaña Aranguren, M.; Wilkinson, M.D.; Egea-Cortines, M. Quantitative Evaluation of Bias in PCR Amplification and Next-Generation Sequencing Derived from Metabarcoding Samples. Anal. Bioanal. Chem. 2015, 407, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Faraci, G.; Murphy, G.; Pilcher, C.; Busch, M.P.; Lee, H.Y. Microdrop Human Immunodeficiency Virus Sequencing for Incidence and Drug Resistance Surveillance. J. Infect. Dis. 2021, 224, 1048–1059. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Sample ID | NCBI Accession Number | Year Sample Collected | Sample Type | RNA Number | Viral Load | Approach | Sequencing Platform |
|---|---|---|---|---|---|---|---|---|
| Patient 1 | 2003-P1-S1 | ON228409 | 2003 | serum | 5171 | 2.4 × 106 | Amplicon | Illumina |
| Patient 2 | 2008-P2-S1 | ON228408 | 2008 | serum | R1672 | 1.7 × 109 | Amplicon | Illumina |
| 2008-P2-S2 | ON228429 | Metagenomic | Illumina | |||||
| Patient 3 | 2008-P3-S1 | ON228433 | 2008 | serum | R932 | 4.1 × 106 | Amplicon | ONT |
| 2008-P3-S2 | ON228428 | Metagenomic | Illumina | |||||
| Patient 4 | 2009-P4-S1 | ON228422 | 2009 | serum | R2582 | 5.4 × 104 | Amplicon | Illumina |
| Patient 5 | 2009-P5-S1 | ON228415 | 2009 | serum | R5799 | 1.1 × 105 | Amplicon | Illumina |
| Patient 6 | 2010-P6-S1 | ON228416 | 2010 | serum | R2758 | 2.6 × 105 | Amplicon | Illumina |
| Patient 7 | 2012-P7-S1 | ON228425 | 2012 | serum | R4987 | 6.6 × 105 | Amplicon | Illumina |
| Patient 8 | 2013-P8-S1 | ON228418 | 2013 | serum | R5685 | 6.6 × 105 | Amplicon | Illumina |
| Patient 9 | 2013-P9-S1 | ON228417 | 2013 | serum | R5837 | 6.8 × 105 | Amplicon | Illumina |
| Patient 10 | 2013-P10-S1 | ON228419 | 2013 | serum | R5843 | 4.5 × 103 | Amplicon | Illumina |
| Patient 11 | 2014-P11-S1 | ON228424 | 2014 | blood | NK6645 | 5.2 × 103 | Amplicon | Illumina |
| Patient 12 | 2015-P12-S1 | ON228423 | 2015 | blood | NK8556 | 9.4 × 103 | Amplicon | Illumina |
| Patient 13 | 2017-P13-S1 | ON228426 | 2017 | blood | NK8558 | 8.9 × 102 | Amplicon | Illumina |
| Patient 14 | 2018-P14-S1 | ON228413 | 2018 | blood | NK8563 | 4.5 × 103 | Amplicon | Illumina |
| Patient 15 | 2018-P15-S1 | ON228421 | 2018 | blood | NK8568 | 5.6 × 103 | Amplicon | Illumina |
| Patient 16 | 2018-P16-S1 | ON228427 | 2018 | blood | NK2696 | 3.9 × 104 | Amplicon | Illumina |
| Patient 17 | 2019-P17-S1 | ON228411 | 2019 | blood | 1812 | 5.6 × 103 | Amplicon | Illumina |
| Patient 18 | 2019-P18-S1 | ON228414 | 2019 | blood | NK4357 | 3.4 × 104 | Amplicon | Illumina |
| 2019-P18-S2 | ON228431 | Metagenomic | Illumina | |||||
| Patient 19 | 2020-P19-S1 | ON228434 | 2020 | blood | 11599 | 9.0 × 104 | Amplicon | ONT |
| 2020-P19-S2 | ON228412 | Amplicon | Illumina | |||||
| 2020-P19-S3 | ON228432 | Metagenomic | Illumina | |||||
| Patient 20 | 2020-P20-S1 | ON228420 | 2020 | blood | 12340 | 5.1 × 103 | Amplicon | Illumina |
| Patient 21 | 2020-P21-S1 | ON228410 | 2020 | blood | NK6108 | 1.2 × 105 | Amplicon | Illumina |
| 2020-P21-S2 | ON228430 | Metagenomic | Illumina |
| Length of Sequence | Length of Polyprotein | Length of UTR (5′ URT/3′ UTR) [bp] | GC Content | |||||
|---|---|---|---|---|---|---|---|---|
| Ampli | Meta | Ampli | Meta | Ampli | Meta | Ampli | Meta | |
| Reference NC_001672.1 | 11,141 | 10,245 bp /3414 aa | 131/764 | 54% | ||||
| 2008-P2-S1/S2 | 11,142 | 11,091 | 10,245 bp /3414 aa | 10,245 bp /3414 aa | 131/766 | 131/715 | 54% | 54% |
| 2019-P18-S1/S2 | 11,009 | 11,005 | 10,245 bp /3414 aa | 10,245 bp /3414 aa | 87/673 | 87/670 | 53% | 53% |
| 2020-P19-S1/S2 | 11,008 | 11,041 | 10,245 bp /3414 aa | 10,245 bp /3414 aa | 73/730 | 106/690 | 54% | 54% |
| 2020-P21-S1/S2 | 11,009 | 11,057 | 10,245 bp /3414 aa | 10,245 bp /3414 aa | 87/677 | 111/701 | 54% | 54% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakotnik, S.; Knap, N.; Bogovič, P.; Zorec, T.M.; Poljak, M.; Strle, F.; Avšič-Županc, T.; Korva, M. Complete Genome Sequencing of Tick-Borne Encephalitis Virus Directly from Clinical Samples: Comparison of Shotgun Metagenomic and Targeted Amplicon-Based Sequencing. Viruses 2022, 14, 1267. https://doi.org/10.3390/v14061267
Zakotnik S, Knap N, Bogovič P, Zorec TM, Poljak M, Strle F, Avšič-Županc T, Korva M. Complete Genome Sequencing of Tick-Borne Encephalitis Virus Directly from Clinical Samples: Comparison of Shotgun Metagenomic and Targeted Amplicon-Based Sequencing. Viruses. 2022; 14(6):1267. https://doi.org/10.3390/v14061267
Chicago/Turabian StyleZakotnik, Samo, Nataša Knap, Petra Bogovič, Tomaž Mark Zorec, Mario Poljak, Franc Strle, Tatjana Avšič-Županc, and Miša Korva. 2022. "Complete Genome Sequencing of Tick-Borne Encephalitis Virus Directly from Clinical Samples: Comparison of Shotgun Metagenomic and Targeted Amplicon-Based Sequencing" Viruses 14, no. 6: 1267. https://doi.org/10.3390/v14061267
APA StyleZakotnik, S., Knap, N., Bogovič, P., Zorec, T. M., Poljak, M., Strle, F., Avšič-Županc, T., & Korva, M. (2022). Complete Genome Sequencing of Tick-Borne Encephalitis Virus Directly from Clinical Samples: Comparison of Shotgun Metagenomic and Targeted Amplicon-Based Sequencing. Viruses, 14(6), 1267. https://doi.org/10.3390/v14061267