
Generated Randomly and Selected Functionally? The Nature of Enterovirus Recombination

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Method
2.1. Virus and Cell Culture
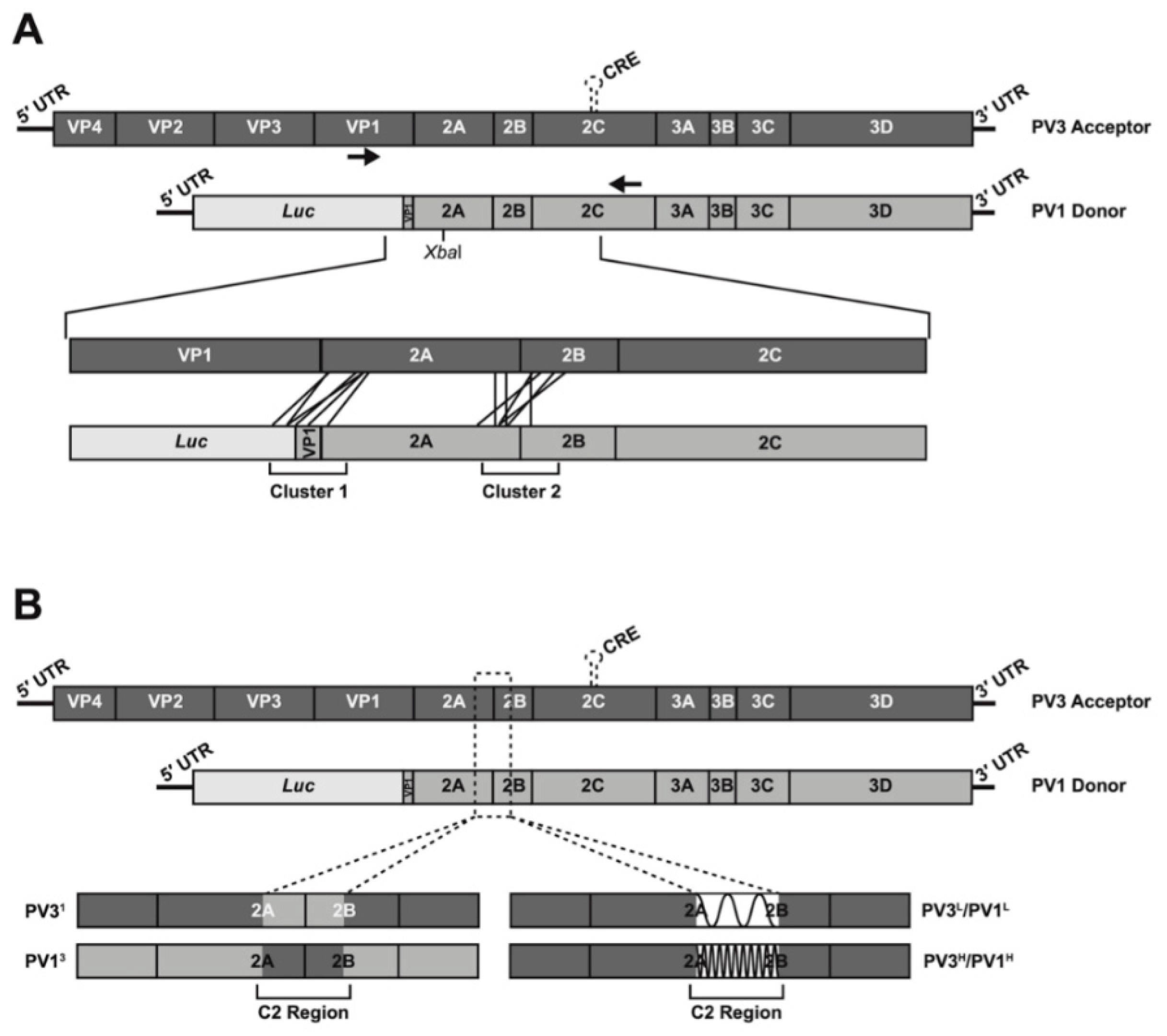
2.2. Design of Modified 2A/2B Junction Sequences
2.3. CRE-REP Assay, Plasmids, In Vitro RNA Transcription and Transfection
2.4. RNA Extraction and RT-PCR
2.5. NGS Library Preparation
2.6. Nomenclature
3. Results
3.1. Influence of RNA Structure and Sequence on CRE-REP Recombination
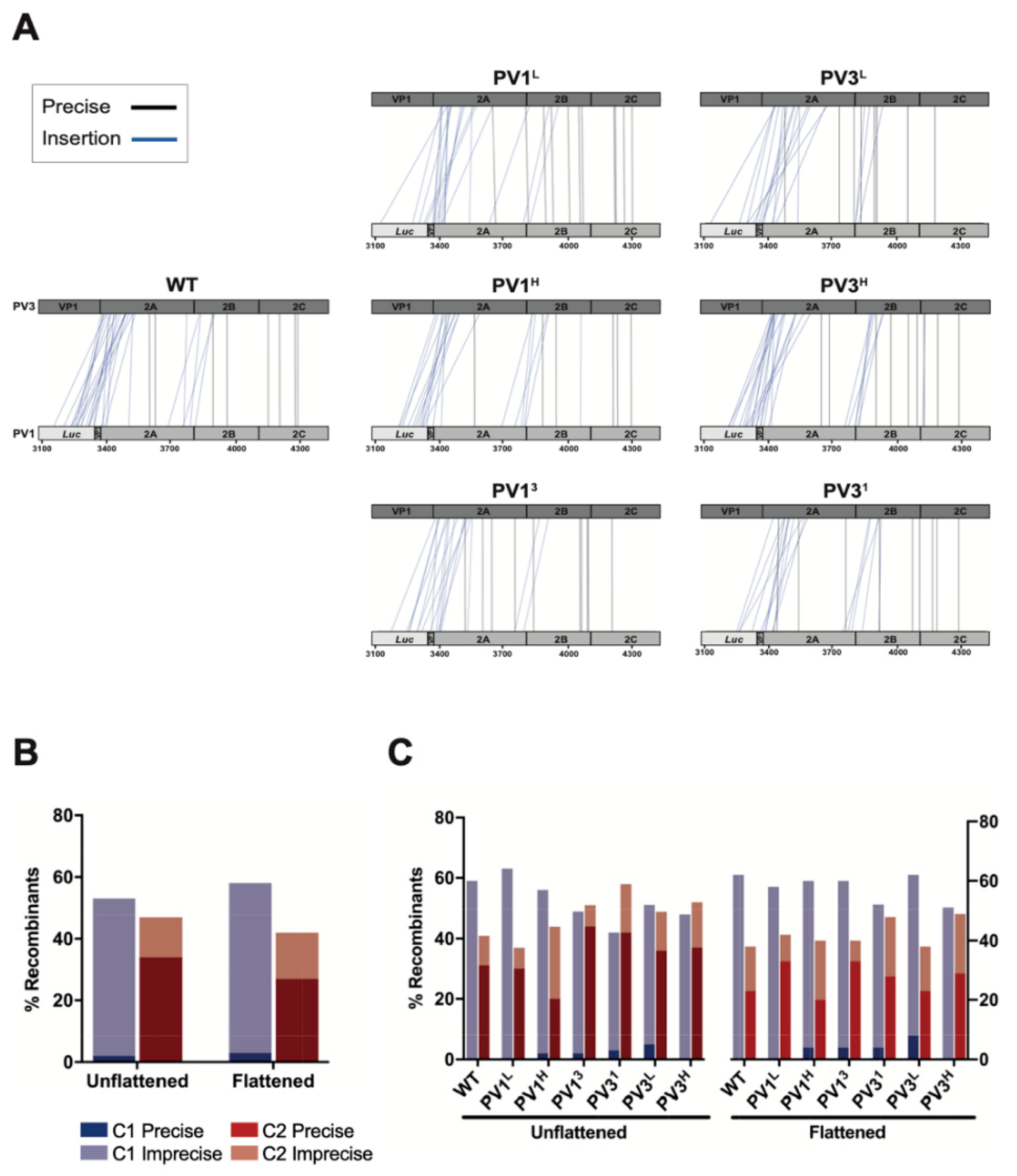
3.2. Analysis of Recombination Junctions in the Modified CRE-REP Assay
3.3. Isolation of Recombinants Following Virus Coinfection
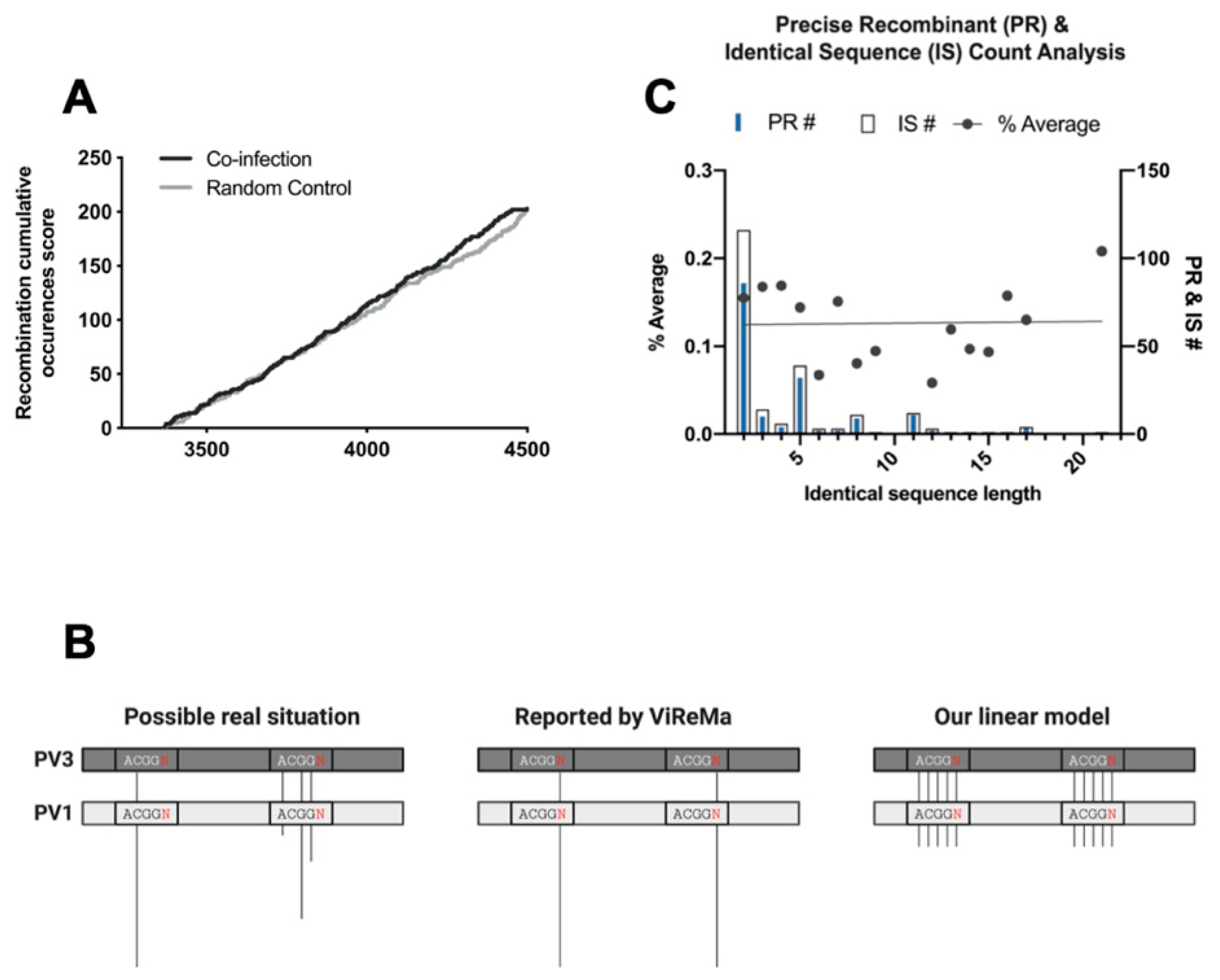
3.4. The Complexity of Recombination Events Revealed by NGS Sequence Analysis
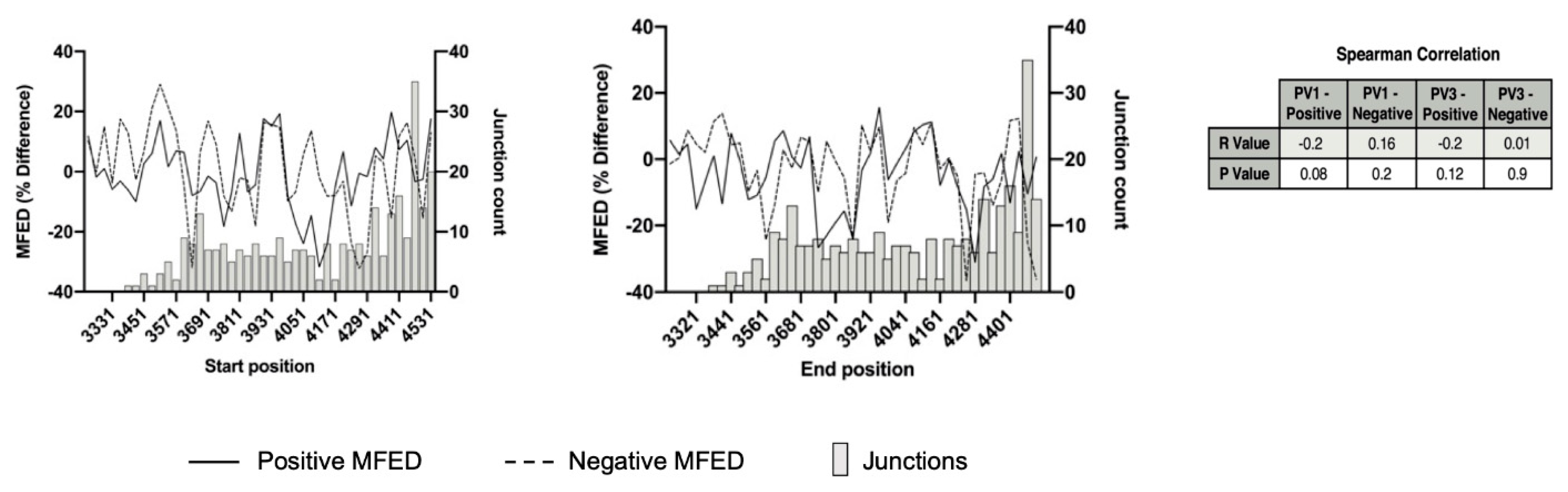
3.5. Analysis of Recombination Junctions Following NGS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hahn, C.S.; Lustig, S.; Strauss, E.G.; Strauss, J.H. Western equine encephalitis virus is a recombinant virus. Proc. Natl. Acad. Sci. USA 1988, 85, 5997–6001. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Li, K.S.; Guan, Y.; Wang, J.; Smith, G.J.; Xu, K.M.; Duan, L.; Rahardjo, A.P.; Puthavathana, P.; Buranathai, C.; Nguyen, T.D.; et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 2004, 430, 209–213. [Google Scholar] [CrossRef]
- Smith, G.J.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [Green Version]
- Gmyl, A.P.; Belousov, E.V.; Maslova, S.V.; Khitrina, E.V.; Chetverin, A.B.; Agol, V.I. Nonreplicative RNA recombination in poliovirus. J. Virol. 1999, 73, 8958–8965. [Google Scholar] [CrossRef] [Green Version]
- Gmyl, A.P.; Korshenko, S.A.; Belousov, E.V.; Khitrina, E.V.; Agol, V.I. Nonreplicative homologous RNA recombination: Promiscuous joining of RNA pieces? RNA 2003, 9, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- King, A.M. Preferred sites of recombination in poliovirus RNA: An analysis of 40 intertypic cross-over sequences. Nucleic Acids Res. 1988, 16, 11705–11723. [Google Scholar] [CrossRef] [Green Version]
- Kirkegaard, K.; Baltimore, D. The mechanism of RNA recombination in poliovirus. Cell 1986, 47, 433–443. [Google Scholar] [CrossRef]
- Woodman, A.; Arnold, J.J.; Cameron, C.E.; Evans, D.J. Biochemical and genetic analysis of the role of the viral polymerase in enterovirus recombination. Nucleic Acids Res. 2016, 44, 6883–6895. [Google Scholar] [CrossRef]
- Nagy, P.D.; Bujarski, J.J. Efficient system of homologous RNA recombination in brome mosaic virus: Sequence and structure requirements and accuracy of crossovers. J. Virol. 1995, 69, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Sergiescu, D.; Aubert-Combiescu, A.; Crainic, R. Recombination between guanidine-resistant and dextran sulfate-resistant mutants of type 1 poliovirus. J. Virol. 1969, 3, 326–330. [Google Scholar] [CrossRef] [Green Version]
- White, K.A.; Morris, T.J. RNA determinants of junction site selection in RNA virus recombinants and defective interfering RNAs. RNA 1995, 1, 1029–1040. [Google Scholar]
- Nagy, P.D.; Zhang, C.; Simon, A.E. Dissecting RNA recombination in vitro: Role of RNA sequences and the viral replicase. EMBO J. 1998, 17, 2392–2403. [Google Scholar] [CrossRef]
- Carpenter, C.D.; Oh, J.W.; Zhang, C.; Simon, A.E. Involvement of a stem-loop structure in the location of junction sites in viral RNA recombination. J. Mol. Biol. 1995, 245, 608–622. [Google Scholar] [CrossRef]
- Bruyere, A.; Wantroba, M.; Flasinski, S.; Dzianott, A.; Bujarski, J.J. Frequent homologous recombination events between molecules of one RNA component in a multipartite RNA virus. J. Virol. 2000, 74, 4214–4219. [Google Scholar] [CrossRef] [Green Version]
- Bujarski, J.J.; Kaesberg, P. Genetic recombination between RNA components of a multipartite plant virus. Nature 1986, 321, 528–531. [Google Scholar] [CrossRef]
- Rowe, C.L.; Fleming, J.O.; Nathan, M.J.; Sgro, J.Y.; Palmenberg, A.C.; Baker, S.C. Generation of coronavirus spike deletion variants by high-frequency recombination at regions of predicted RNA secondary structure. J. Virol. 1997, 71, 6183–6190. [Google Scholar] [CrossRef] [Green Version]
- Lowry, K.; Woodman, A.; Cook, J.; Evans, D.J. Recombination in enteroviruses is a biphasic replicative process involving the generation of greater-than genome length ’imprecise’ intermediates. PLoS Pathog. 2014, 10, e1004191. [Google Scholar] [CrossRef]
- Simmonds, P. SSE: A nucleotide and amino acid sequence analysis platform. BMC Res. Notes 2012, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, P.; Tuplin, A.; Evans, D.J. Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: Implications for virus evolution and host persistence. RNA 2004, 10, 1337–1351. [Google Scholar] [CrossRef] [Green Version]
- Egger, D.; Bienz, K. Intracellular location and translocation of silent and active poliovirus replication complexes. J. Gen. Virol. 2005, 86, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Bowtie 2: Manual. Available online: http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml#bowtie2-options-score-min (accessed on 1 April 2022).
- Routh, A.; Johnson, J.E. Discovery of functional genomic motifs in viruses with ViReMa-a Virus Recombination Mapper-for analysis of next-generation sequencing data. Nucleic Acids Res. 2014, 42, e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alnaji, F.G.; Holmes, J.R.; Rendon, G.; Vera, J.C.; Fields, C.J.; Martin, B.E.; Brooke, C.B. Sequencing Framework for the Sensitive Detection and Precise Mapping of Defective Interfering Particle-Associated Deletions across Influenza A and B Viruses. J. Virol. 2019, 93, e00354-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figlerowicz, M. Role of RNA structure in non-homologous recombination between genomic molecules of brome mosaic virus. Nucleic Acids Res. 2000, 28, 1714–1723. [Google Scholar] [CrossRef] [Green Version]
- Cascone, P.J.; Haydar, T.F.; Simon, A.E. Sequences and structures required for recombination between virus-associated RNAs. Science 1993, 260, 801–805. [Google Scholar] [CrossRef]
- Bentley, K.; Alnaji, F.G.; Woodford, L.; Jones, S.; Woodman, A.; Evans, D.J. Imprecise recombinant viruses evolve via a fitness-driven, iterative process of polymerase template-switching events. PLoS Pathog. 2021, 17, e1009676. [Google Scholar] [CrossRef]
- Davis, M.; Sagan, S.M.; Pezacki, J.P.; Evans, D.J.; Simmonds, P. Bioinformatic and physical characterizations of genome-scale ordered RNA structure in mammalian RNA viruses. J. Virol. 2008, 82, 11824–11836. [Google Scholar] [CrossRef] [Green Version]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef]
- Worobey, M.; Holmes, E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999, 80 Pt 10, 2535–2543. [Google Scholar] [CrossRef]
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [Green Version]
- Kempf, B.J.; Watkins, C.L.; Peersen, O.B.; Barton, D.J. Picornavirus RNA Recombination Counteracts Error Catastrophe. J. Virol. 2019, 93, e00652-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotty, S.; Cameron, C.E.; Andino, R. RNA virus error catastrophe: Direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA 2001, 98, 6895–6900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentley, K.; Evans, D.J. Mechanisms and consequences of positive-strand RNA virus recombination. J. Gen. Virol. 2018, 99, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses 2019, 11, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Negative Control | Co-Infection | Technical Replicate | |
|---|---|---|---|
| Total reads | 797,386.00 | 1,488,344.00 | 1,023,536.00 |
| Aligned to PV1 | 11,754.00 | 279,958.00 | 46,028.00 |
| Aligned to PV3 | 525,214.00 | 1,111,733.00 | 909,263.00 |
| Not Mapped * | 260,418.00 | 69,292.00 | 61,749.00 |
| Recombination reads | 0.00 | 27,361.00 | 6496.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alnaji, F.G.; Bentley, K.; Pearson, A.; Woodman, A.; Moore, J.; Fox, H.; Macadam, A.J.; Evans, D.J. Generated Randomly and Selected Functionally? The Nature of Enterovirus Recombination. Viruses 2022, 14, 916. https://doi.org/10.3390/v14050916
Alnaji FG, Bentley K, Pearson A, Woodman A, Moore J, Fox H, Macadam AJ, Evans DJ. Generated Randomly and Selected Functionally? The Nature of Enterovirus Recombination. Viruses. 2022; 14(5):916. https://doi.org/10.3390/v14050916
Chicago/Turabian StyleAlnaji, Fadi G., Kirsten Bentley, Ashley Pearson, Andrew Woodman, Jonathan Moore, Helen Fox, Andrew J. Macadam, and David J. Evans. 2022. "Generated Randomly and Selected Functionally? The Nature of Enterovirus Recombination" Viruses 14, no. 5: 916. https://doi.org/10.3390/v14050916
APA StyleAlnaji, F. G., Bentley, K., Pearson, A., Woodman, A., Moore, J., Fox, H., Macadam, A. J., & Evans, D. J. (2022). Generated Randomly and Selected Functionally? The Nature of Enterovirus Recombination. Viruses, 14(5), 916. https://doi.org/10.3390/v14050916