
Comparative Proteomic Analysis Reveals Mx1 Inhibits Senecavirus A Replication in PK-15 Cells by Interacting with the Capsid Proteins VP1, VP2 and VP3

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus and Antibodies
2.2. 50% Tissue Culture Infectious Dose (TCID50) Assay
2.3. Indirect Immunofluorescence Assay (IFA)
2.4. Protein Sample Preparation, Trypsin Digestion and TMT Labeling
2.5. High-pH Reversed-Phase Liquid Chromatography Fractionation
2.6. LC-MS/MS Analysis
2.7. Data Availability
2.8. Database Search
2.9. Bioinformatics Analysis
2.10. Quantitative Real-Time PCR (qPCR)
2.11. Plasmid Construction
2.12. Western Blot (WB)
2.13. RNA Interference
2.14. Co-Immunoprecipitation (Co-IP) and Confocal Immunofluorescence Microscopy
2.15. GTPase Activity Assay
2.16. Statistical Analysis
3. Results
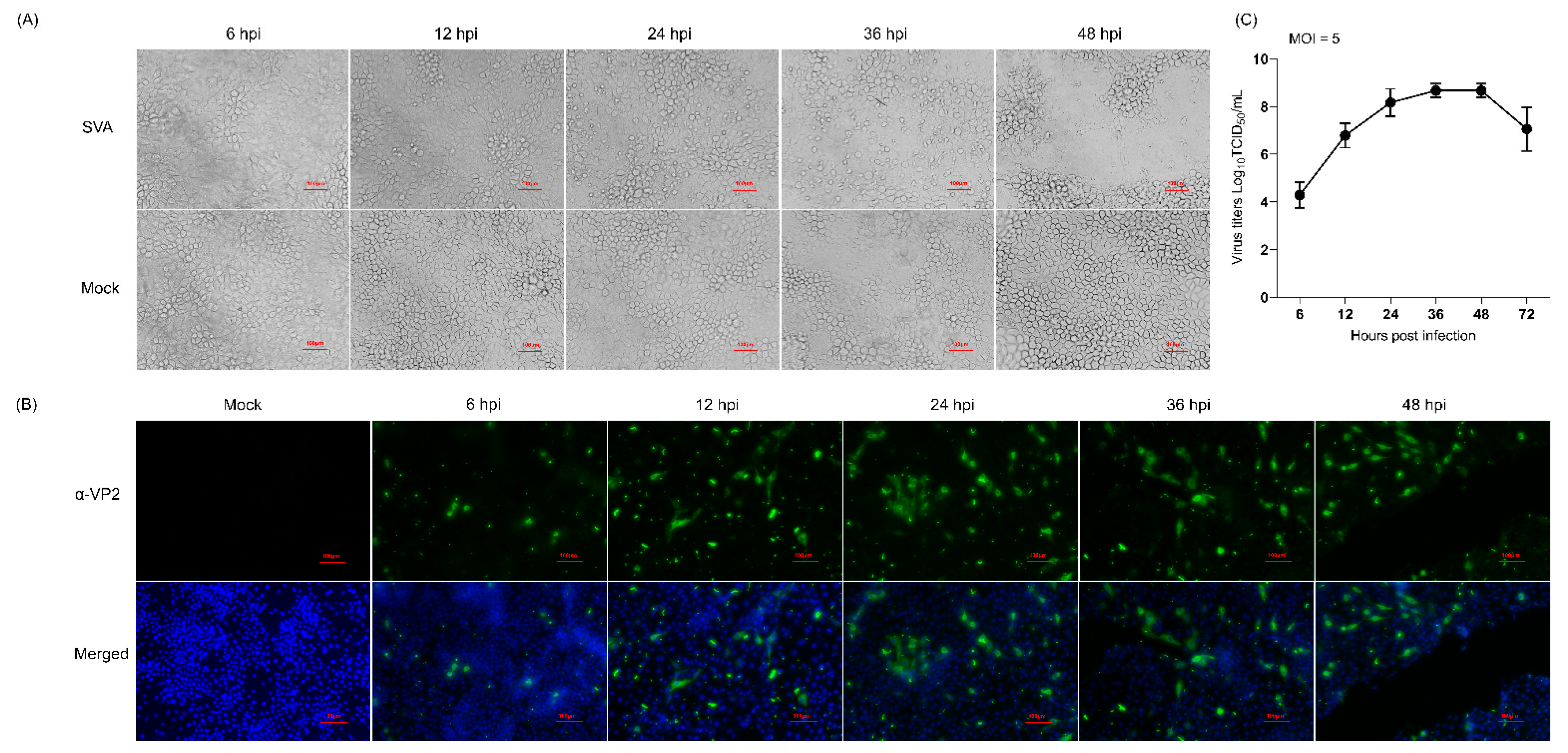
3.1. Determination of the Optimal Time Points for Proteomic Analysis Following SVA Infection
3.2. Temporal Proteomic Analysis of PK-15 Cells in Response to SVA Infection
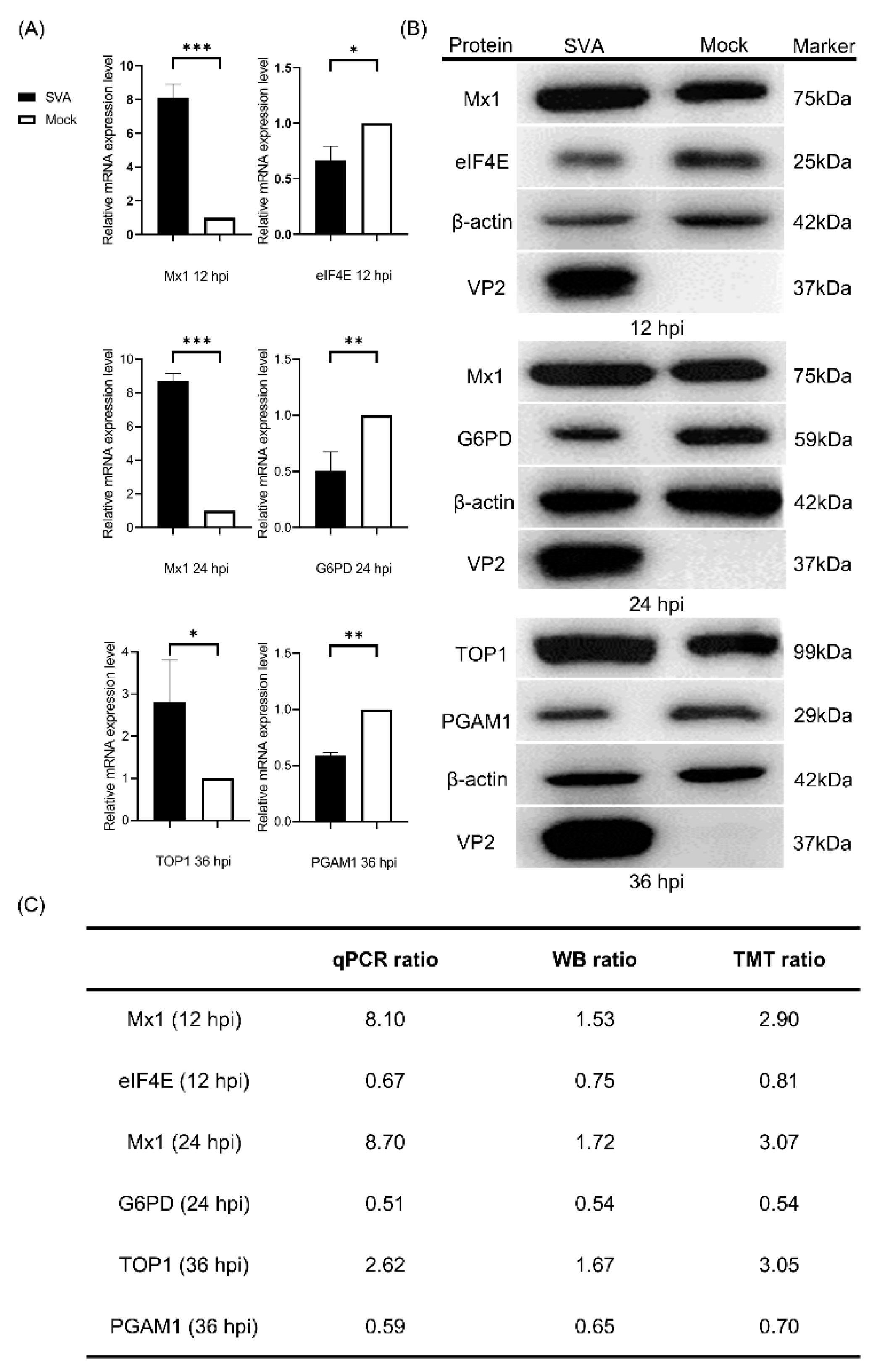
3.3. Validation of the Proteomics Data by qPCR and WB
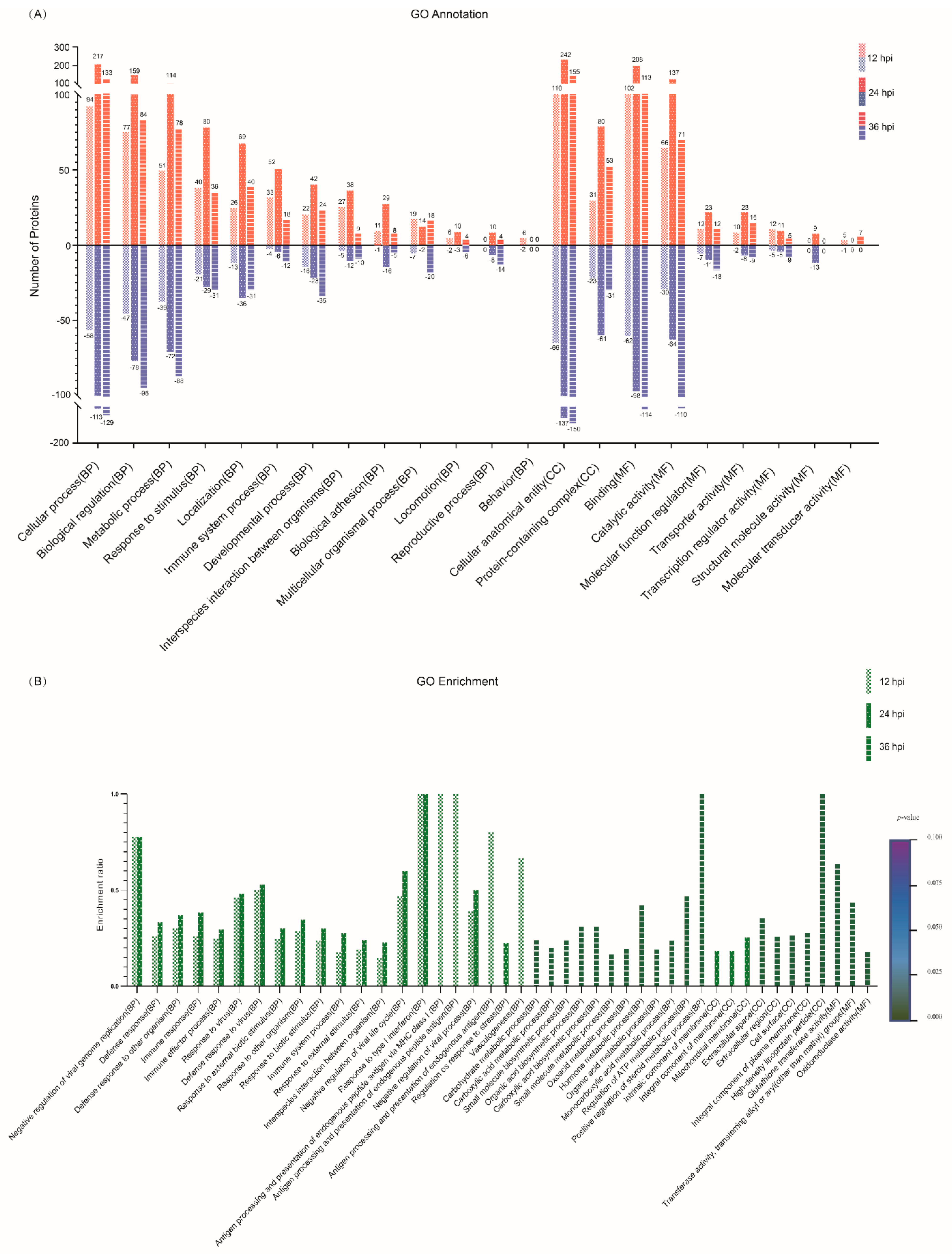
3.4. GO Functional Annotation of the DEPs
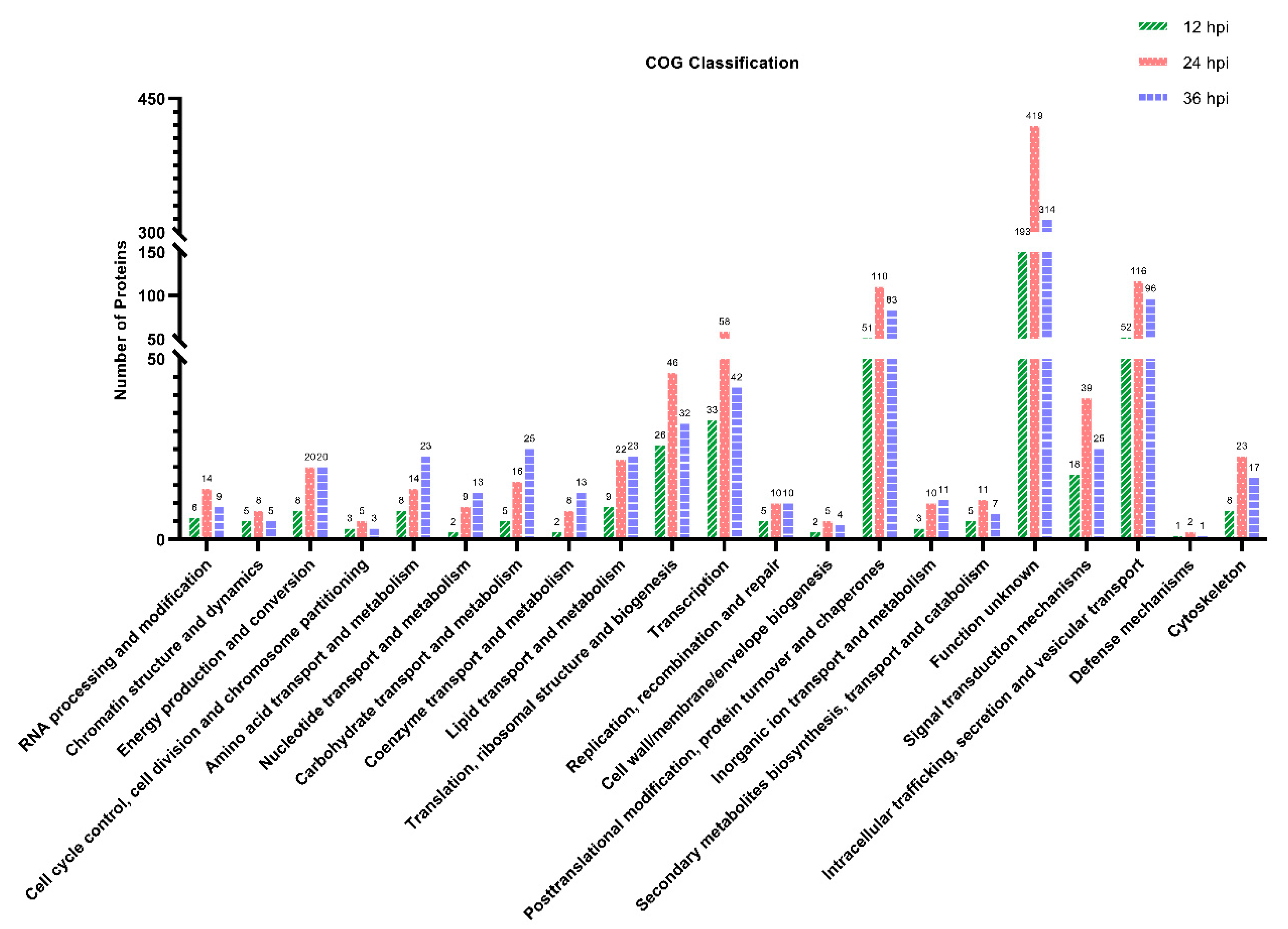
3.5. COG Functional Classification of the DEPs
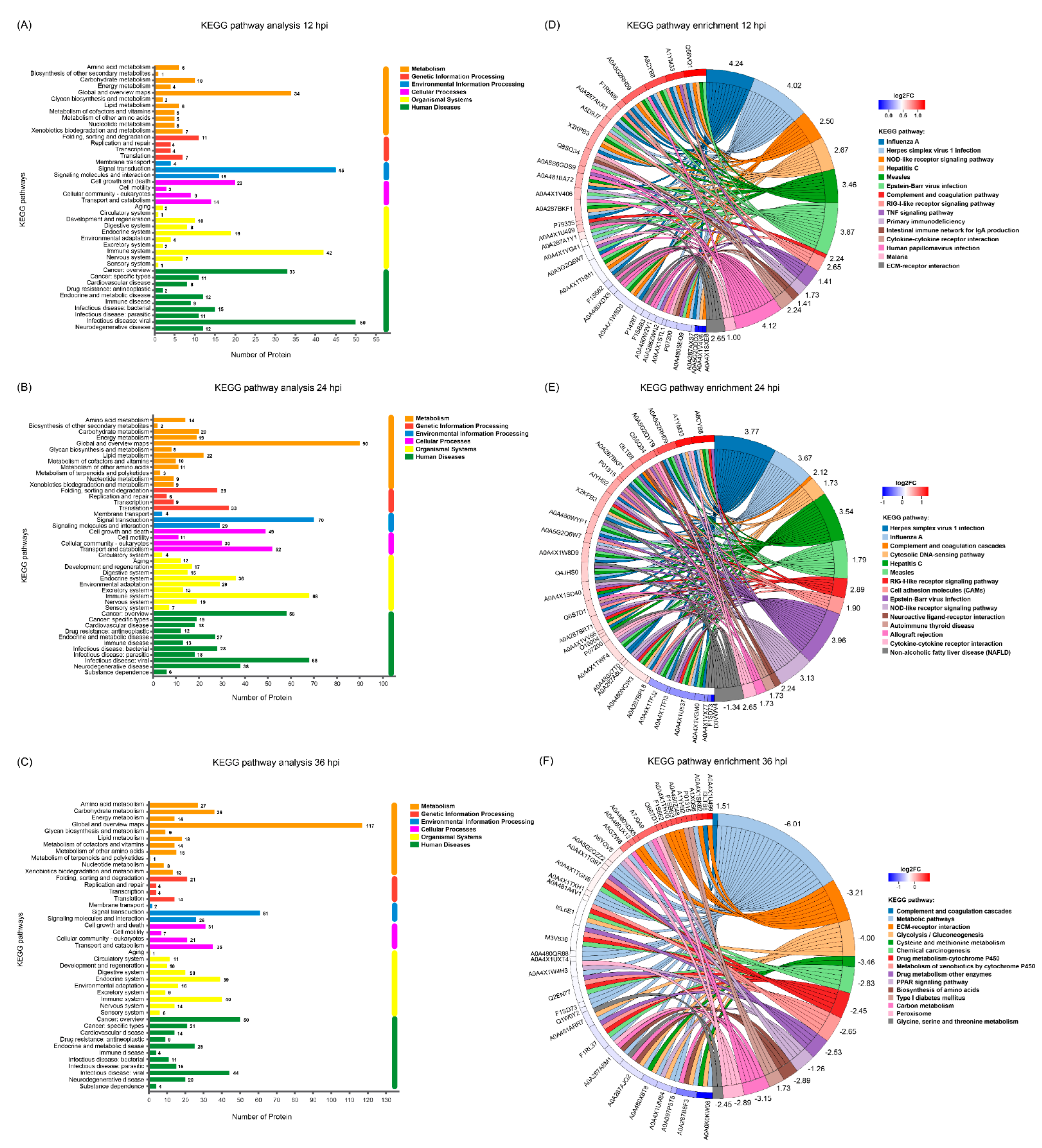
3.6. KEGG Pathway Analysis of the DEPs
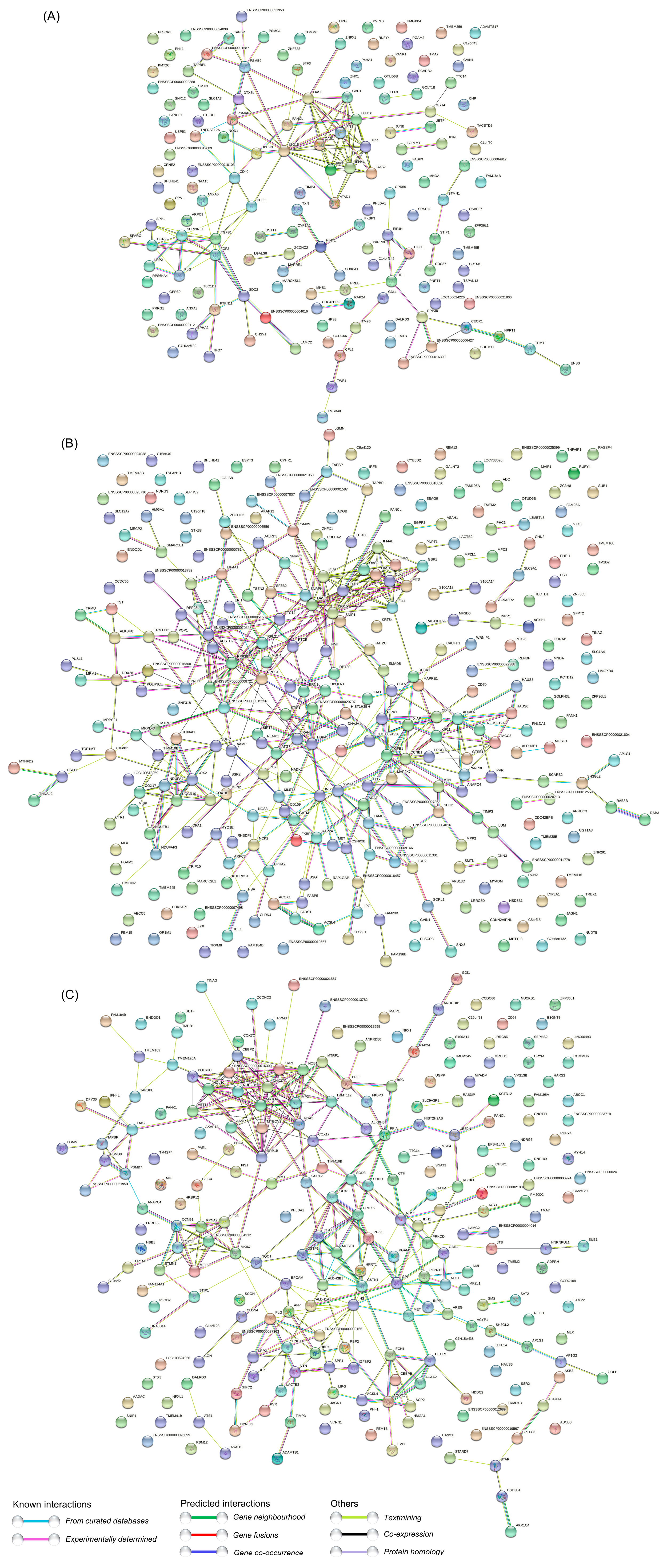
3.7. PPI Network Analysis of the DEPs
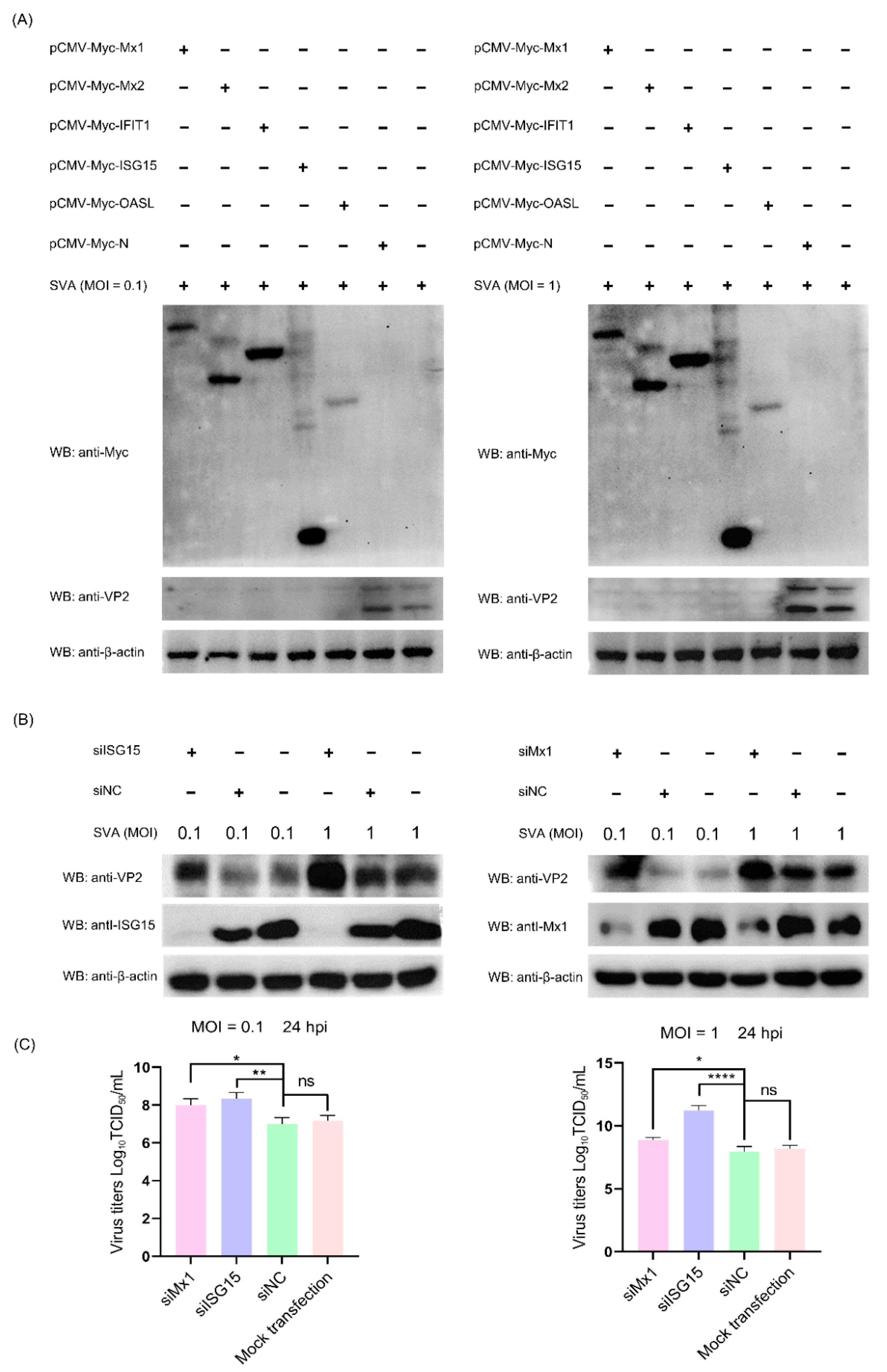
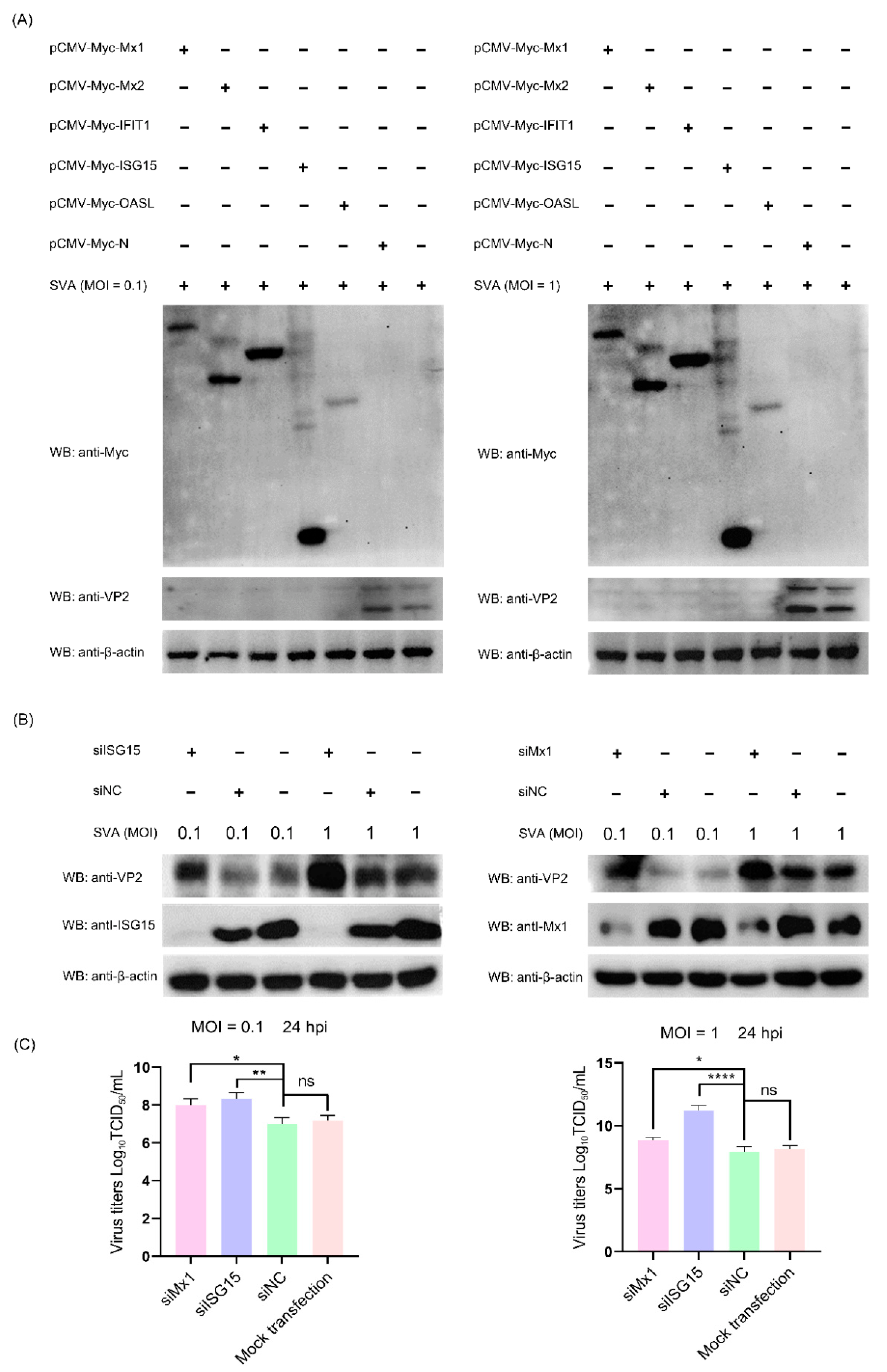
3.8. The ISG Family Proteins Inhibit SVA Replication
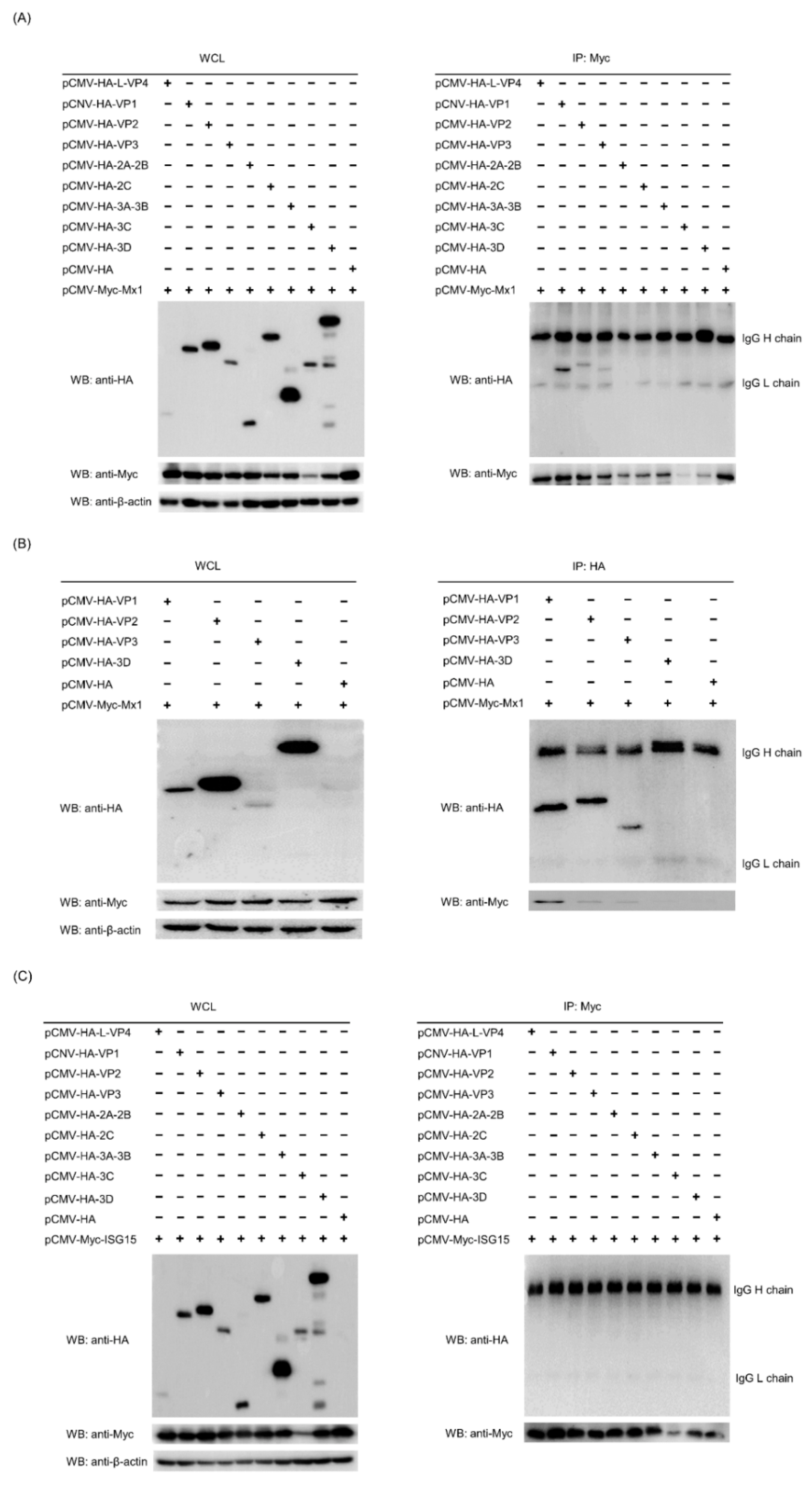
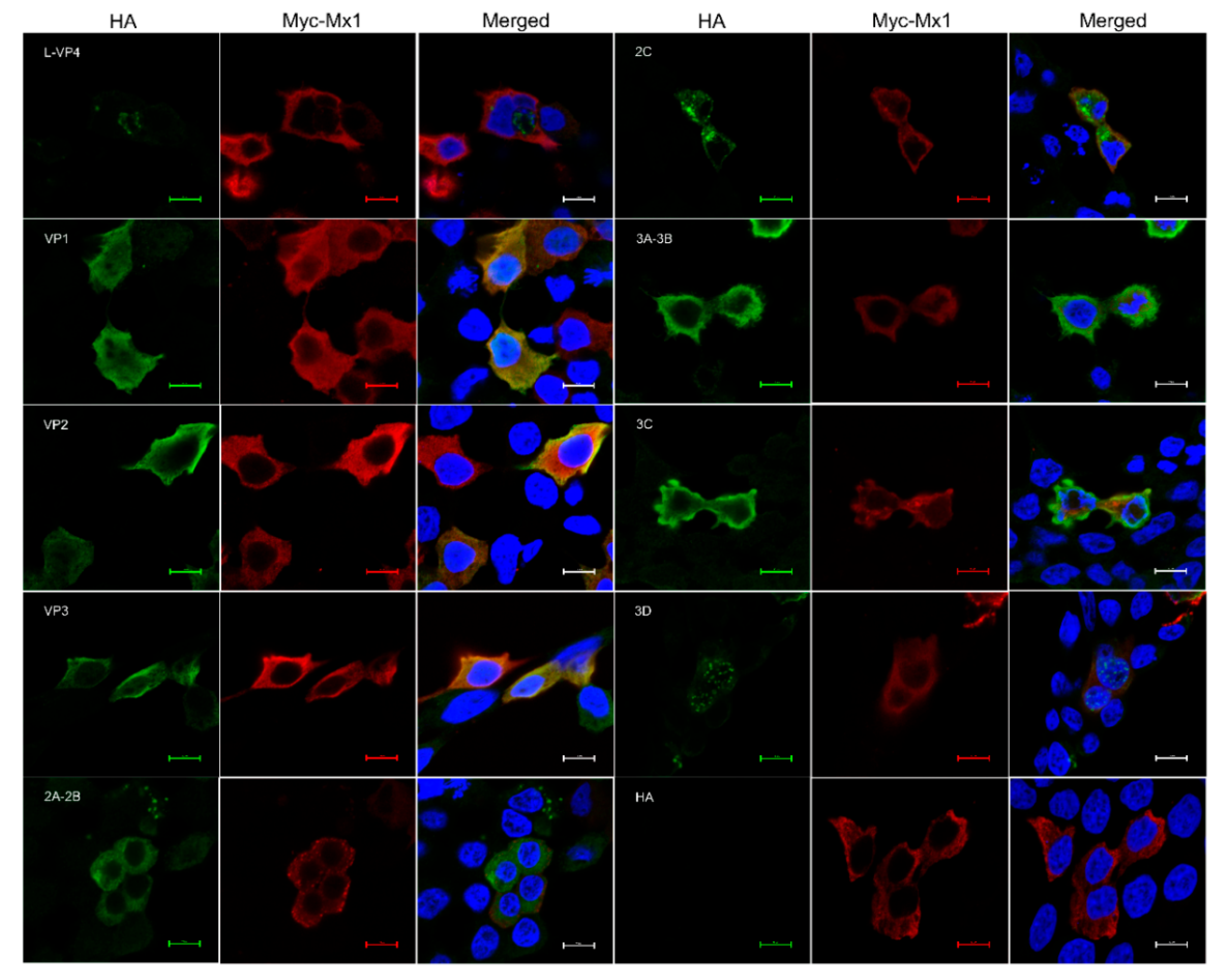
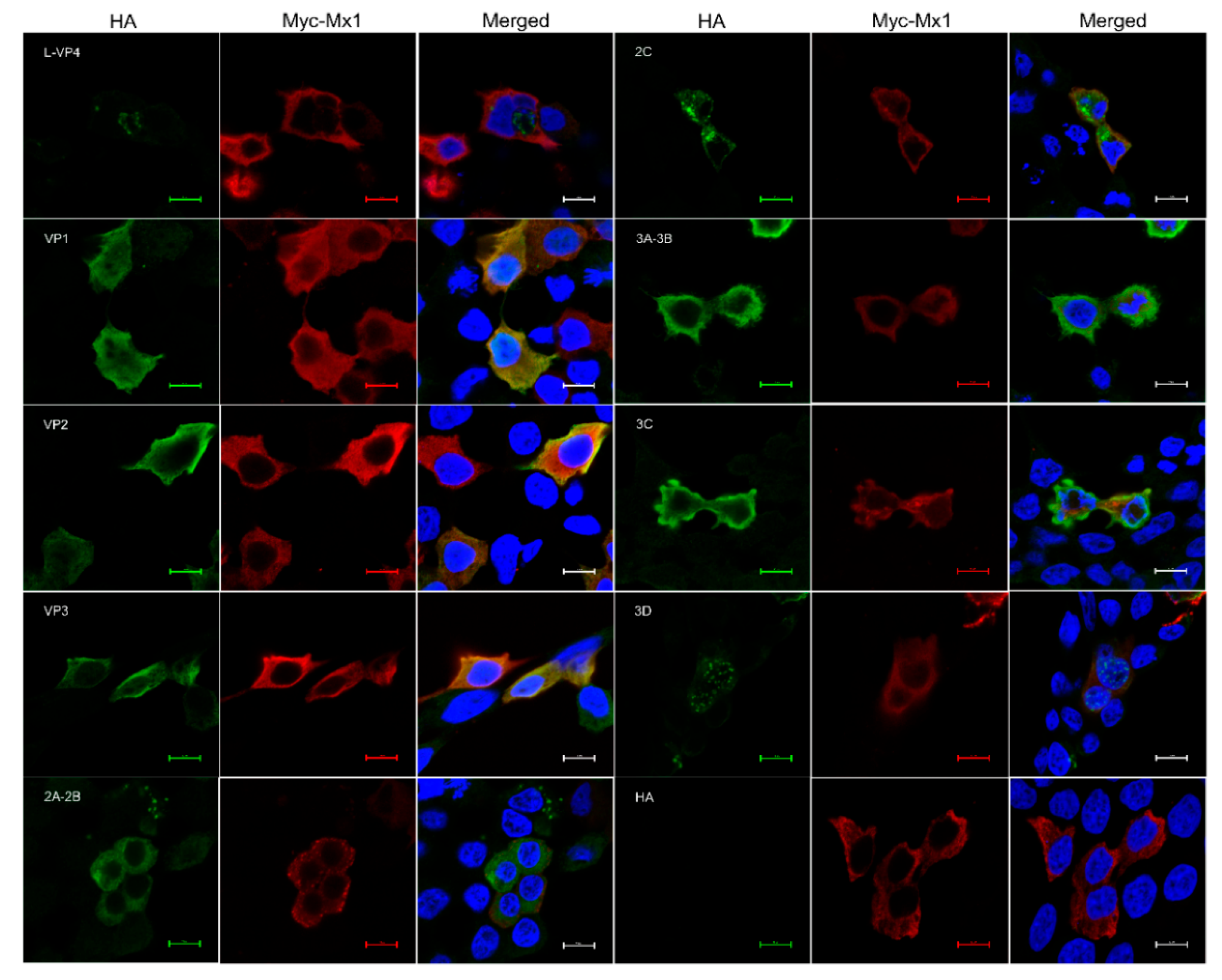
3.9. Mx1 but Not ISG15 Interacts with SVA VP1, VP2 and VP3 Proteins
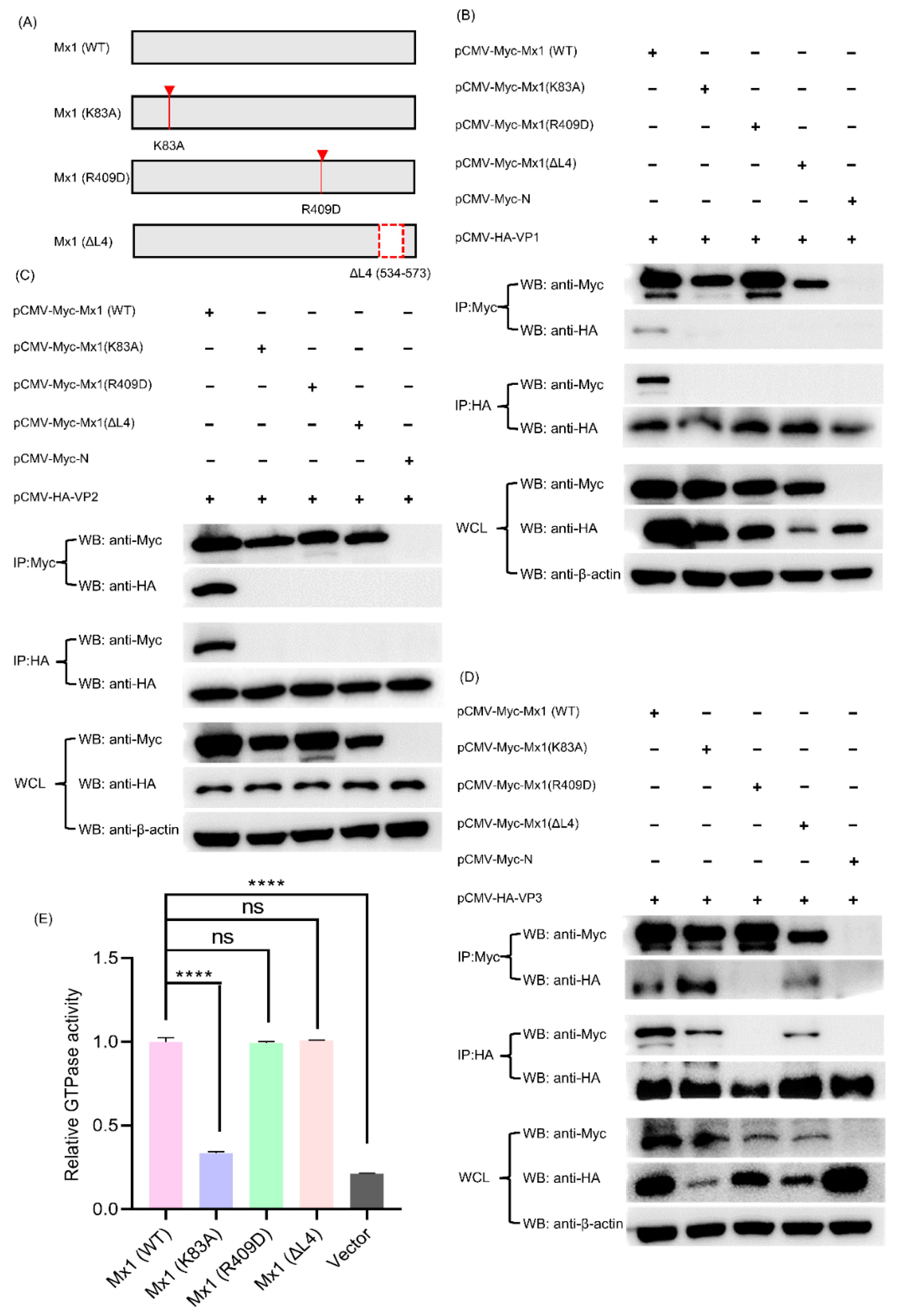
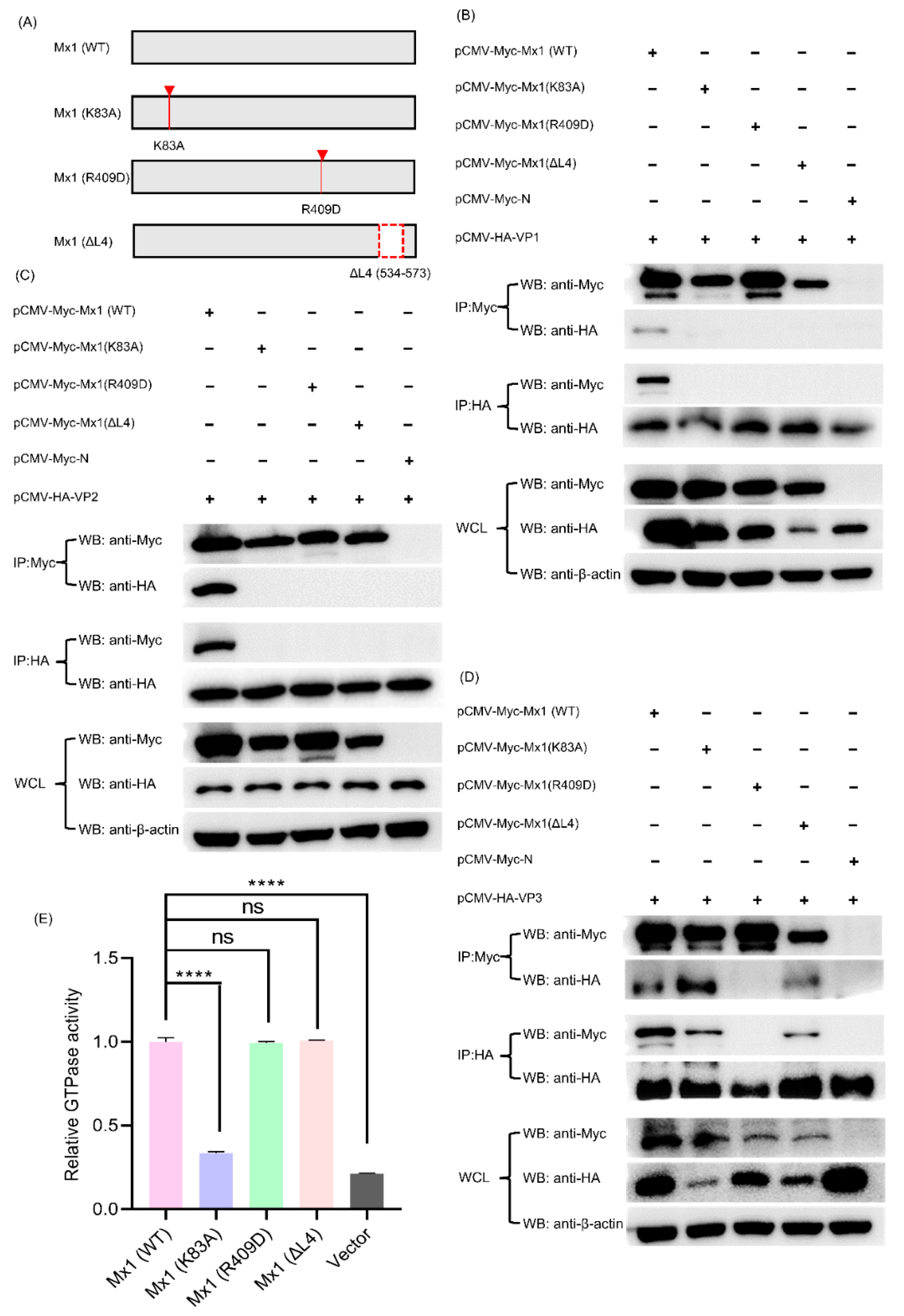
3.10. Mx1-Associated GTPase, Oligomerization and Interaction Activities Are Required for Its Interaction with VP1, VP2 and VP3 Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hales, L.M.; Knowles, N.J.; Reddy, P.S.; Xu, L.; Hay, C.; Hallenbeck, P.L. Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J. Gen. Virol. 2008, 89 Pt 5, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Pasma, T.; Davidson, S.; Shaw, S.L. Idiopathic vesicular disease in swine in Manitoba. Can. Vet. J. 2008, 49, 84–85. [Google Scholar] [PubMed]
- Knowles, N.J.; Hales, L.M.; Jones, B.H.; Landgraf, J.G.; House, J.A.; Skele, K.L.; Burroughs, K.D.; Hallenbeck, P.L. Epidemiology of Seneca Valley virus: Identification and characterization of isolates from pigs in the United States. In Proceedings of the Abstracts of the Northern Lights EUROPIC 2006—XIV Meeting of the European Study Group on the Molecular Biology of Picornaviruses, Saariselkä, Finland, 26 November–1 December 2006. Abstract G2. [Google Scholar]
- Vannucci, F.A.; Linhares, D.C.; Barcellos, D.E.; Lam, H.C.; Collins, J.; Marthaler, D. Identification and complete genome of Seneca Valley Virus in vesicular fluid and sera of pigs affected with idiopathic vesicular disease, Brazil. Transbound. Emerg. Dis. 2015, 62, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Montiel, N.; Buckley, A.; Guo, B.; Kulshreshtha, V.; VanGeelen, A.; Hoang, H.; Rademacher, C.; Yoon, K.J.; Lager, K. Vesicular disease in 9-week-old pigs experimentally infected with Senecavirus A. Emerg. Infect. Dis. 2016, 22, 1246–1248. [Google Scholar] [CrossRef] [Green Version]
- Canning, P.; Canon, A.; Bates, J.L.; Gerardy, K.; Linhares, D.C.; Piñeyro, P.E.; Schwartz, K.J.; Yoon, K.J.; Rademacher, C.J.; Holtkamp, D.; et al. Neonatal mortality, vesicular lesions and lameness associated with Senecavirus A in a U.S. sow farm. Transbound. Emerg. Dis. 2016, 63, 373–378. [Google Scholar] [CrossRef]
- Segalés, J.; Barcellos, D.; Alfieri, A.; Burrough, E.; Marthaler, D. Senecavirus A: An emerging pathogen causing vesicular disease and mortality in pigs. Vet. Pathol. 2017, 54, 11–21. [Google Scholar] [CrossRef]
- Dall Agnol, A.M.; Miyabe, F.M.; Leme, R.A.; Oliveira, T.E.S.; Headley, S.A.; Alfieri, A.A.; Alfieri, A.F. Quantitative analysis of Senecavirus A in tissue samples from naturally infected newborn piglets. Arch. Virol. 2018, 163, 527–531. [Google Scholar] [CrossRef]
- Maggioli, M.F.; Fernandes, M.H.V.; Joshi, L.R.; Sharma, B.; Tweet, M.M.; Noll, J.C.G.; Bauermann, F.V.; Diel, D.G. Persistent infection and transmission of Senecavirus A from carrier sows to contact piglets. J. Virol. 2019, 93, e00819-19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, F.; Li, K.; Cao, W.; Ru, Y.; Chen, S.; Li, S.; Liu, X.; Zhu, Z.; Zheng, H. The insufficient activation of RIG-I-like signaling pathway contributes to highly efficient replication of porcine picornaviruses in IBRS-2 Cells. Mol. Cell. Proteom. 2021, 20, 100147. [Google Scholar] [CrossRef]
- Houston, E.; Temeeyasen, G.; Piñeyro, P.E. Comprehensive review on immunopathogenesis, diagnostic and epidemiology of Senecavirus A. Virus Res. 2020, 286, 198038. [Google Scholar] [CrossRef]
- Li, P.; Zhang, X.; Cao, W.; Yang, F.; Du, X.; Shi, Z.; Zhang, M.; Liu, X.; Zhu, Z.; Zheng, H. RIG-I is responsible for activation of type I interferon pathway in Seneca Valley virus-infected porcine cells to suppress viral replication. Virol. J. 2018, 15, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Q.; Liu, H.; Zhu, Z.; Yang, F.; Ma, L.; Cai, X.; Xue, Q.; Zheng, H. Seneca Valley virus 3Cpro abrogates the IRF3- and IRF7-mediated innate immune response by degrading IRF3 and IRF7. Virology 2018, 518, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hou, L.; Quan, R.; Wang, D.; Jiang, H.; Liu, J. Synergetic contributions of viral VP1, VP3, and 3C to activation of the AKT-AMPK-MAPK-MTOR signaling pathway for Seneca Valley Virus-induced autophagy. J. Virol. 2022, 96, e0155021. [Google Scholar] [CrossRef] [PubMed]
- Pappireddi, N.; Martin, L.; Wühr, M. A review on quantitative multiplexed proteomics. ChemBioChem 2019, 20, 1210–1224. [Google Scholar] [CrossRef]
- Klann, K.; Tascher, G.; Münch, C. Virus systems biology: Proteomics profiling of dynamic protein networks during infection. Adv. Virus Res. 2021, 109, 1–29. [Google Scholar] [CrossRef]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and their applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhou, L.; Ge, X.; Han, J.; Guo, X.; Chen, Y.; Zhang, Y.; Yang, H. Development of a VP2-based real-time fluorescent reverse transcription recombinase-aided amplification assay to rapidly detect Senecavirus A. Transbound. Emerg. Dis. 2021, 1–12. [Google Scholar] [CrossRef]
- Ramesh, A.K.; Parreño, V.; Schmidt, P.J.; Lei, S.; Zhong, W.; Jiang, X.; Emelko, M.B.; Yuan, L. Evaluation of the 50% infectious dose of human Norovirus cin-2 in gnotobiotic pigs: A comparison of classical and contemporary methods for endpoint estimation. Viruses 2020, 12, 955. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Zhou, X.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Zhang, Y.; Yang, H. Quantitative proteomic analysis of porcine intestinal epithelial cells infected with porcine deltacoronavirus using iTRAQ-Coupled LC-MS/MS. J. Proteome Res. 2020, 19, 4470–4485. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, J.; Zhang, X.M.; Gao, Z.C.; Liu, C.C.; Zhang, Y.N.; Hou, J.X.; Li, Z.Y.; Kan, L.; Li, W.L.; et al. Porcine Mx1 protein inhibits Classical Swine Fever virus replication by targeting nonstructural protein NS5B. J. Virol. 2018, 92, e02147-17. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Li, C.; Dong, L.; Yang, G.; Liu, H. Tandem mass tag-based quantitative proteomic analysis of ISG15 knockout PK15 cells in pseudorabies virus infection. Genes 2021, 12, 1557. [Google Scholar] [CrossRef]
- Verhelst, J.; Parthoens, E.; Schepens, B.; Fiers, W.; Saelens, X. Interferon-inducible protein Mx1 inhibits influenza virus by interfering with functional viral ribonucleoprotein complex assembly. J. Virol. 2012, 86, 13445–13455. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Sridharan, H.; Chen, R.; Baker, D.P.; Wang, S.; Krug, R.M. Influenza B virus non-structural protein 1 counteracts ISG15 antiviral activity by sequestering ISGylated viral proteins. Nat. Commun. 2016, 7, 12754. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Wang, Z.; Chen, J.; Li, H.; Lin, Z.; Zhang, F.; Zhou, Y.; Hou, J. GTPase activity is not essential for the interferon-inducible MxA protein to inhibit the replication of hepatitis B virus. Arch. Virol. 2008, 153, 1677–1684. [Google Scholar] [CrossRef]
- Verhelst, J.; Spitaels, J.; Nurnberger, C.; De Vlieger, D.; Ysenbaert, T.; Staeheli, P.; Fiers, W.; Saelens, X. Functional comparison of Mx1 from two different mouse species reveals the involvement of loop L4 in the antiviral activity against influenza A viruses. J. Virol. 2015, 89, 10879–10890. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhao, X.; Bai, Y.; Sun, B.; Xie, Q.; Ma, J. The first identification and complete genome of Senecavirus A affecting pig with idiopathic vesicular disease in China. Transbound. Emerg. Dis. 2017, 64, 1633–1640. [Google Scholar] [CrossRef]
- Fernandes, M.H.V.; Maggioli, M.F.; Joshi, L.R.; Clement, T.; Faccin, T.C.; Rauh, R.; Bauermann, F.V.; Diel, D.G. Pathogenicity and cross-reactive immune responses of a historical and a contemporary Senecavirus A strains in pigs. Virology 2018, 522, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.C.; Michael, D.D.; Faaberg, K.S.; Guo, B.; Yoon, K.J.; Lager, K.M. Comparison of historical and contemporary isolates of Senecavirus A. Vet. Microbiol. 2021, 253, 108946. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Kong, N.; Dong, S.; Chen, X.; Qin, W.; Wang, H.; Jiao, Y.; Zhai, H.; Li, L.; Gao, F.; et al. 2AB protein of Senecavirus A antagonizes selective autophagy and type I interferon production by degrading LC3 and MARCHF8. Autophagy 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Li, X.; Yin, M.; Wang, H.; Qin, L.; Li, H.; Liu, W.; Zhao, Z.; Zhao, Q.; Chen, H.; et al. Selective autophagy receptor SQSTM1/ p62 inhibits Seneca Valley virus replication by targeting viral VP1 and VP3. Autophagy 2021, 17, 3763–3775. [Google Scholar] [CrossRef]
- Liu, T.; Li, X.; Wu, M.; Qin, L.; Chen, H.; Qian, P. Seneca Valley virus 2C and 3Cpro induce apoptosis via mitochondrion-mediated intrinsic pathway. Front. Microbiol. 2019, 10, 1202. [Google Scholar] [CrossRef]
- Nie, X.; Qian, L.; Sun, R.; Huang, B.; Dong, X.; Xiao, Q.; Zhang, Q.; Lu, T.; Yue, L.; Chen, S.; et al. Multi-organ proteomic landscape of COVID-19 autopsies. Cell 2021, 184, 775–791.e14. [Google Scholar] [CrossRef]
- Leme, R.A.; Oliveira, T.E.S.; Alfieri, A.F.; Headley, S.A.; Alfieri, A.A. Pathological, immunohistochemical and molecular findings associated with Senecavirus A-induced lesions in neonatal piglets. J. Comp. Pathol. 2016, 155, 145–155. [Google Scholar] [CrossRef]
- McKendrick, L.; Pain, V.M.; Morley, S.J. Translation initiation factor 4E. Int. J. Biochem. Cell Biol. 1999, 31, 31–35. [Google Scholar] [CrossRef]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Champoux, J.J. DNA topoisomerase I-mediated nicking of circular duplex DNA. Methods Mol. Biol. 2001, 95, 81–87. [Google Scholar] [CrossRef]
- Sharif, F.; Rasul, A.; Ashraf, A.; Hussain, G.; Younis, T.; Sarfraz, I.; Chaudhry, M.A.; Bukhari, S.A.; Ji, X.Y.; Selamoglu, Z.; et al. Phosphoglycerate mutase 1 in cancer: A promising target for diagnosis and therapy. IUBMB Life 2019, 71, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bai, J.; Fan, H.; Yan, J.; Li, S.; Jiang, P. E2 ubiquitin-conjugating enzyme UBE2L6 promotes Senecavirus A proliferation by stabilizing the viral RNA polymerase. PLoS Pathog. 2020, 16, e1008970. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mou, C.; Wang, M.; Pan, S.; Chen, Z. Transcriptome analysis of Senecavirus A-infected cells: Type I interferon is a critical anti-viral factor. Microb. Pathog. 2020, 147, 104432. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ni, B.; Wei, R. Comparative proteomic profiling: Cellular metabolisms are mainly affected in Senecavirus A-inoculated cells at an early stage of infection. Viruses 2021, 13, 1036. [Google Scholar] [CrossRef]
- Qian, S.; Fan, W.; Liu, T.; Wu, M.; Zhang, H.; Cui, X.; Zhou, Y.; Hu, J.; Wei, S.; Chen, H.; et al. Seneca Valley virus suppresses host type I interferon production by targeting adaptor proteins MAVS, TRIF, and TANK for cleavage. J. Virol. 2017, 91, e00823-17. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Liu, H.; Zhu, Z.; Yang, F.; Xue, Q.; Cai, X.; Liu, X.; Zheng, H. Seneca Valley virus 3C protease negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. Antivir. Res. 2018, 160, 183–189. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmids Name | Primers | Sequence (5′-3′) | Restriction Enzymes | GenBank Accession Numbers |
|---|---|---|---|---|
| pCMV-Myc-Mx1 | pCMV-Myc-Mx1-F | GCCATGGAGGCCCGAATTCGGATGGTTTATTCCAACTGTGAAAG | EcoR I | DQ095779.1 |
| pCMV-Myc-Mx1-R | GCGGCCGCGGTACCTCGAGTCAGCCTGGGAACTTGG | Xho I | ||
| pCMV-Myc-Mx2 | pCMV-Myc-Mx2-F | GCCATGGAGGCCCGAATTCGGATGCCTAAACCCCGCAT | EcoR I | NM_001097416.1 |
| pCMV-Myc-Mx2-R | GCGGCCGCGGTACCTCGAGTTACATCCCTTGTACCTCAACC | Xho I | ||
| pCMV-Myc-ISG15 | pCMV-Myc-ISG15-F | GCCATGGAGGCCCGAATTCGGATGGGTAGGGAACTGAAGGT | EcoR I | EU647216.1 |
| pCMV-Myc-ISG15-R | GCGGCCGCGGTACCTCGAGCTAGCACTCGGTGGGGT | Xho I | ||
| pCMV-Myc-OASL | pCMV-Myc-OASL-F | GCCATGGAGGCCCGAATTCGGATGGAGCTATTTTACACCCCAG | EcoR I | MG679809.1 |
| pCMV-Myc-OASL-R | GCGGCCGCGGTACCTCGAGTCAGTCACAGCCTTTGGCT | Xho I | ||
| pCMV-Myc-IFIT1 | pCMV-Myc-IFIT1-F | GCCATGGAGGCCCGAATTCGGATGAGTAATAATGCTGATGAAGATCAG | EcoR I | JN621781.1 |
| pCMV-Myc-IFIT1-R | GCGGCCGCGGTACCTCGAGTTAGGGATCAAGTCCCTCAGATT | Xho I | ||
| pCMV-HA-L-VP4 | pCMV-HA-L-VP4-F | GCCATGGAGGCCCGAATTCGGATGCAGAACTCTCATTTTTCTTTC | EcoR I | MN433300.1 |
| pCMV-HA-L-VP4-R | GCGGCCGCGGTACCTCGAGTCATTTGAGGTAGCCAAGAGGGTT | Xho I | ||
| pCMV-HA-VP1 | pCMV-HA-VP1-F | GCCATGGAGGCCCGAATTCGGATGTCCACCGACAACGCC | EcoR I | MN433300.1 |
| pCMV-HA-VP1-R | GCGGCCGCGGTACCTCGAGTCATTGCATCAGCATCTTCTGC | Xho I | ||
| pCMV-HA-VP2 | pCMV-HA-VP2-F | GCCATGGAGGCCCGAATTCGGATGGATCACAATACCGAAGAAATGG | EcoR I | MN433300.1 |
| pCMV-HA-VP2-R | GCGGCCGCGGTACCTCGAGTCACTGTTCCTCGTCCGTCC | Xho I | ||
| pCMV-HA-VP3 | pCMV-HA-VP3-F | GCCATGGAGGCCCGAATTCGGATGGGGCCCATTCCCAC | EcoR I | MN433300.1 |
| pCMV-HA-VP3-R | GCGGCCGCGGTACCTCGAGTCAGTGGAACACGTAGGAAGGATT | Xho I | ||
| pCMV-HA-2A-2B | pCMV-HA-2A-2B-F | GCCATGGAGGCCCGAATTCGGATGTCAGGCGACATCGAGAC | EcoR I | MN433300.1 |
| pCMV-HA-2A-2B-R | GCGGCCGCGGTACCTCGAGTCATTGCATCTTGAACAGCTTTC | Xho I | ||
| pCMV-HA-2C | pCMV-HA-2C-F | GCCATGGAGGCCCGAATTCGGATGGGACCCATGGACACAGTC | EcoR I | MN433300.1 |
| pCMV-HA-2C-R | GCGGCCGCGGTACCTCGAGTCACTGTAGAACCAGAGTCTGCATATTTC | Xho I | ||
| pCMV-HA-3A-3B | pCMV-HA-3A-3B-F | GCCATGGAGGCCCGAATTCGGATGAGCCCTAACGAGAACGACG | EcoR I | MN433300.1 |
| pCMV-HA-3A-3B-R | GCGGCCGCGGTACCTCGAGTCATTGCATTTCCATAAGAGAG | Xho I | ||
| pCMV-HA-3C | pCMV-HA-3C-F | GCCATGGAGGCCCGAATTCGGATGCAGCCCAACGTGGACAT | EcoR I | MN433300.1 |
| pCMV-HA-3C-R | GCGGCCGCGGTACCTCGAGTCATTGCATTGTAGCCAGAGGC | Xho I | ||
| pCMV-HA-3D | pCMV-HA-3D-F | GCCATGGAGGCCCGAATTCGGATGGGACTGATGACTGAGCTAGAGC | EcoR I | MN433300.1 |
| pCMV-HA-3D-R | GCGGCCGCGGTACCTCGAGTCAGTCGAACAAGGCCCT | Xho I | ||
| pCMV-Myc-Mx1(K83A) | pCMV-Myc-Mx1(K83A)-F | CAGTTCGGGCGCGAGCTCCGTGCTGGAGGCCCT | DQ095779.1 | |
| pCMV-Myc-Mx1(K83A)-R | AGCACGGAGCTCGCGCCCGAACTCTGGTCCCCGAT | |||
| pCMV-Myc-Mx1(R409D) | pCMV-Myc-Mx1(R409D)-F | TACCAAGATGGATAATGAGTTCTGCAAATGGAGTGC | DQ095779.1 | |
| pCMV-Myc-Mx1(R409D)-R | AGAACTCATTATCCATCTTGGTAAACAGCCGACACT | |||
| pCMV-Myc-Mx1(ΔL4) | pCMV-Myc-Mx1(ΔL4)-F | ATCGTGTACTCCATAGCCGAGATCTTTCAGCAC | DQ095779.1 | |
| pCMV-Myc-Mx1(ΔL4)-R | GGCTATGGAGTACACGATCTGCTCCATTTGGAAC | |||
| qMx1-F | GGCGTGGGAATCAGTCATG | NM_214061.2 | ||
| qMx1-R | AGGAAGGTCTATGAGGGTCAGATCT | |||
| qTOP1-F | GGCCACCAGTGGAAGGA | XM_013993526.2 | ||
| qTOP1-R | CTCGTCCACCACGCCTT | |||
| qeIF4E-F | GCCTGGCTGTGACTACTCAC | XM_005656555.3 | ||
| qeIF4E-R | TCCAATAAGGCACAGCAGTG | |||
| qG6PD-F | CGCAACTCCTACGTGGC | XM_003360515.5 | ||
| qG6PD-R | GCGGATGTTCTTGGTGAC | |||
| qPGAM1-F | TACAAGCTGGTGCTGATCC | XM_003483535.4 | ||
| qPGAM1-R | CTGCACTGAGGTGAAGCAG | |||
| qβ-actin-F | TCCCTGGAGAAGAGCTACGA | XM_003124280.5 | ||
| qβ-actin-R | AGCACCGTGTTGGCGTAGAG |
| siRNAs Name | Sense (5′-3′) | Antisense (5′-3′) | GenBank Accession Numbers | Sources |
|---|---|---|---|---|
| siMx1-216 | CCAAUCACCUGUUACUAAATT | UUUAGUAACAGGUGAUUGGTT | DQ095779.1 | Designed in this study |
| siMx1-732 | GCAAUCAGCCAUACGACAUTT | AUGUCGUAUGGCUGAUUGCTT | Designed in this study | |
| siMx1-1906 | GCACCUGAUUGCCUACCAUTT | AUGGUAGGCAAUCAGGUGCTT | Designed in this study | |
| siISG15 | CUAUGUGCACCGUGUAUAUTT | AUAUACACGGUGCACAUAGTT | EU647216.1 | [25] |
| siNC | UUCUCCGAACGUGUCACGUTT | ACGUGACACGUUCGGAGAATT | N/A | Designed in this study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, H.; Xiang, Z.; Ge, X.; Zhou, L.; Han, J.; Guo, X.; Chen, Y.; Zhang, Y.; Yang, H. Comparative Proteomic Analysis Reveals Mx1 Inhibits Senecavirus A Replication in PK-15 Cells by Interacting with the Capsid Proteins VP1, VP2 and VP3. Viruses 2022, 14, 863. https://doi.org/10.3390/v14050863
Gao H, Xiang Z, Ge X, Zhou L, Han J, Guo X, Chen Y, Zhang Y, Yang H. Comparative Proteomic Analysis Reveals Mx1 Inhibits Senecavirus A Replication in PK-15 Cells by Interacting with the Capsid Proteins VP1, VP2 and VP3. Viruses. 2022; 14(5):863. https://doi.org/10.3390/v14050863
Chicago/Turabian StyleGao, Hang, Zhaoying Xiang, Xinna Ge, Lei Zhou, Jun Han, Xin Guo, Yanhong Chen, Yongning Zhang, and Hanchun Yang. 2022. "Comparative Proteomic Analysis Reveals Mx1 Inhibits Senecavirus A Replication in PK-15 Cells by Interacting with the Capsid Proteins VP1, VP2 and VP3" Viruses 14, no. 5: 863. https://doi.org/10.3390/v14050863
APA StyleGao, H., Xiang, Z., Ge, X., Zhou, L., Han, J., Guo, X., Chen, Y., Zhang, Y., & Yang, H. (2022). Comparative Proteomic Analysis Reveals Mx1 Inhibits Senecavirus A Replication in PK-15 Cells by Interacting with the Capsid Proteins VP1, VP2 and VP3. Viruses, 14(5), 863. https://doi.org/10.3390/v14050863