
Robust AAV Genotyping Based on Genetic Distances in Rep Gene That Are Maintained by Ubiquitous Recombination

 , ,
, ,  and
and
Abstract
:1. Introduction
1.1. AAV Biology
1.2. AAV Gene Therapy
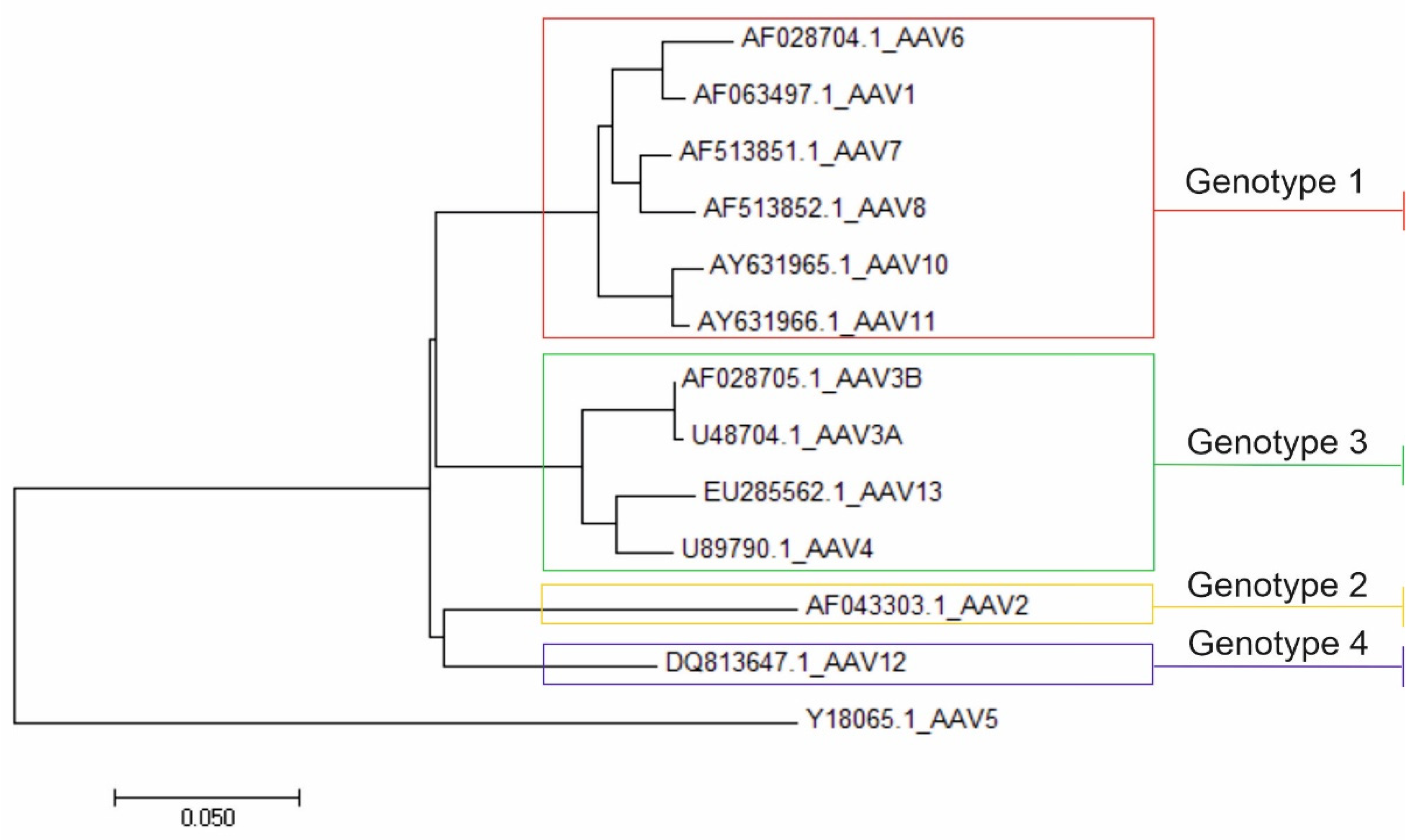
1.3. AAV Classification
2. Materials and Methods
2.1. Sequence Selection for Analysis
2.2. Recombination Analysis
2.3. Nucleotide and Predicted Amino Acid Sequences Analysis
3. Results
3.1. The Dataset
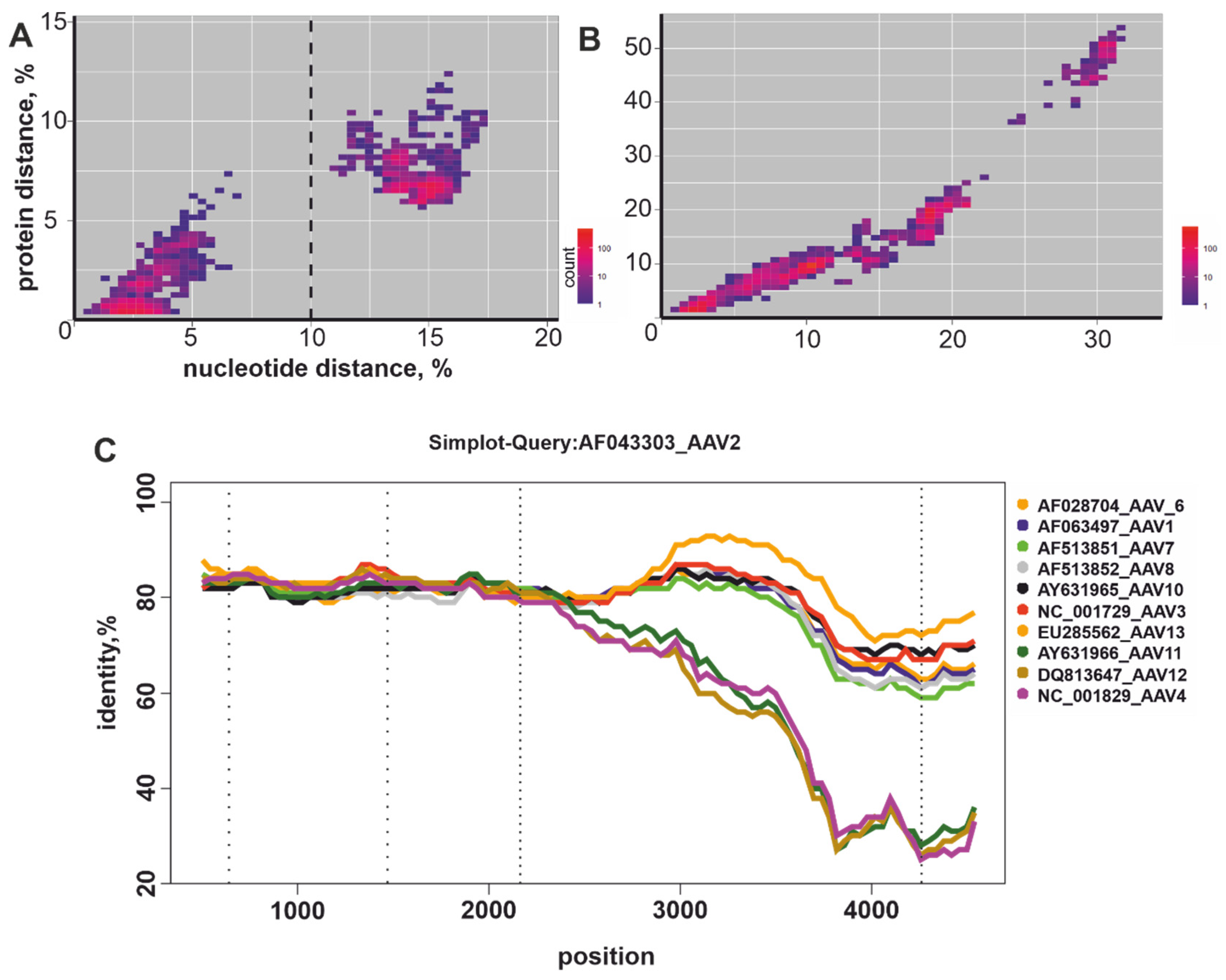
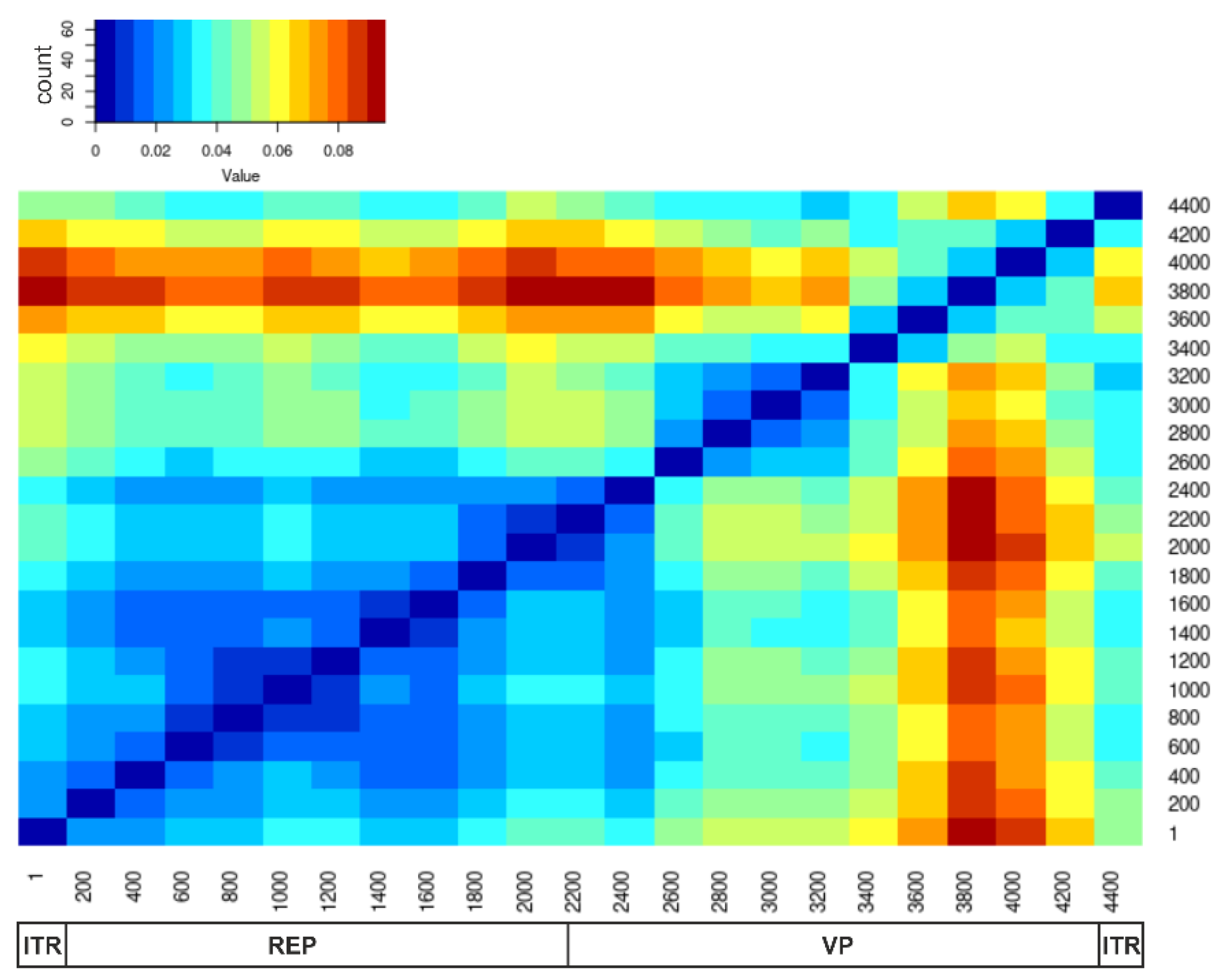
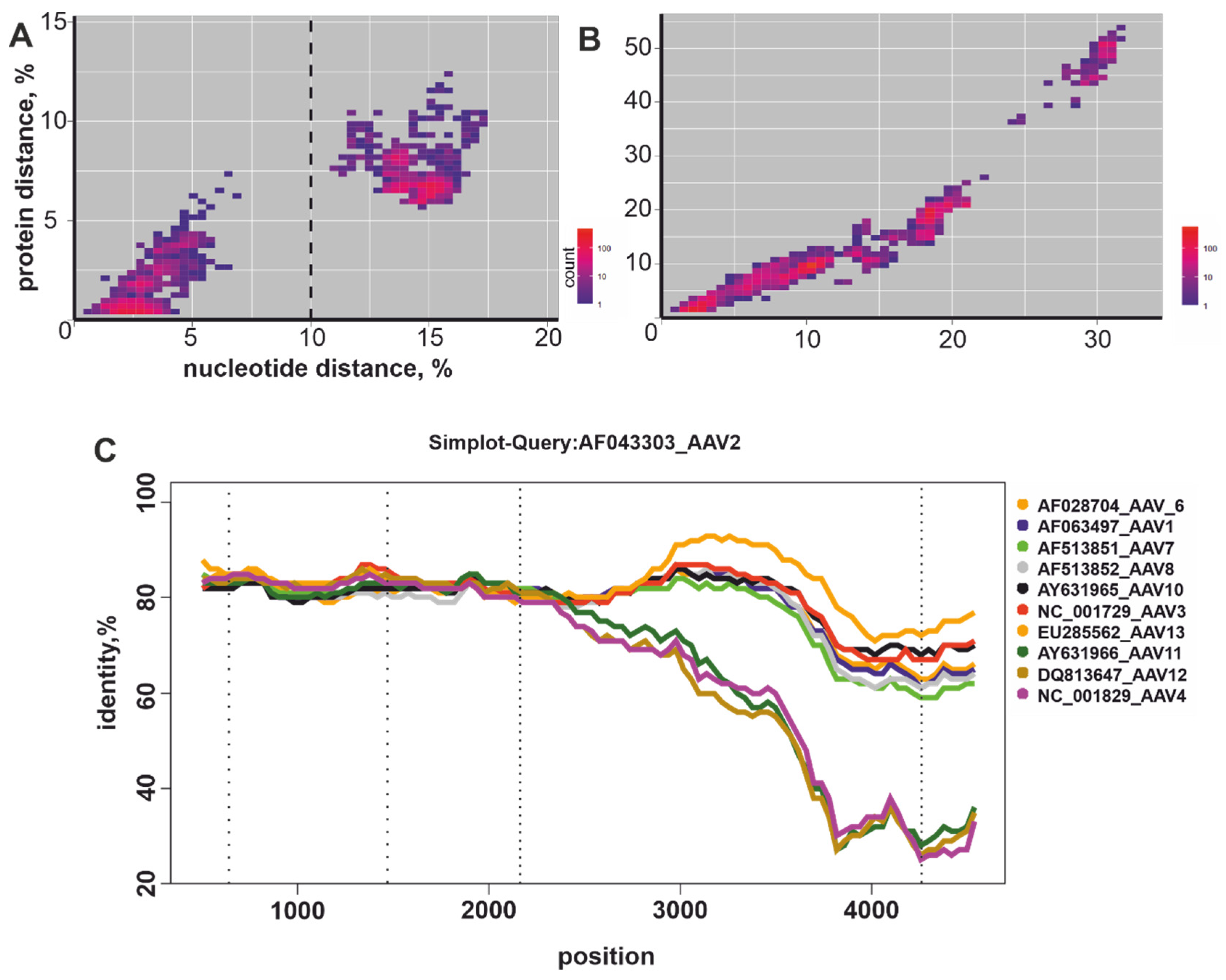
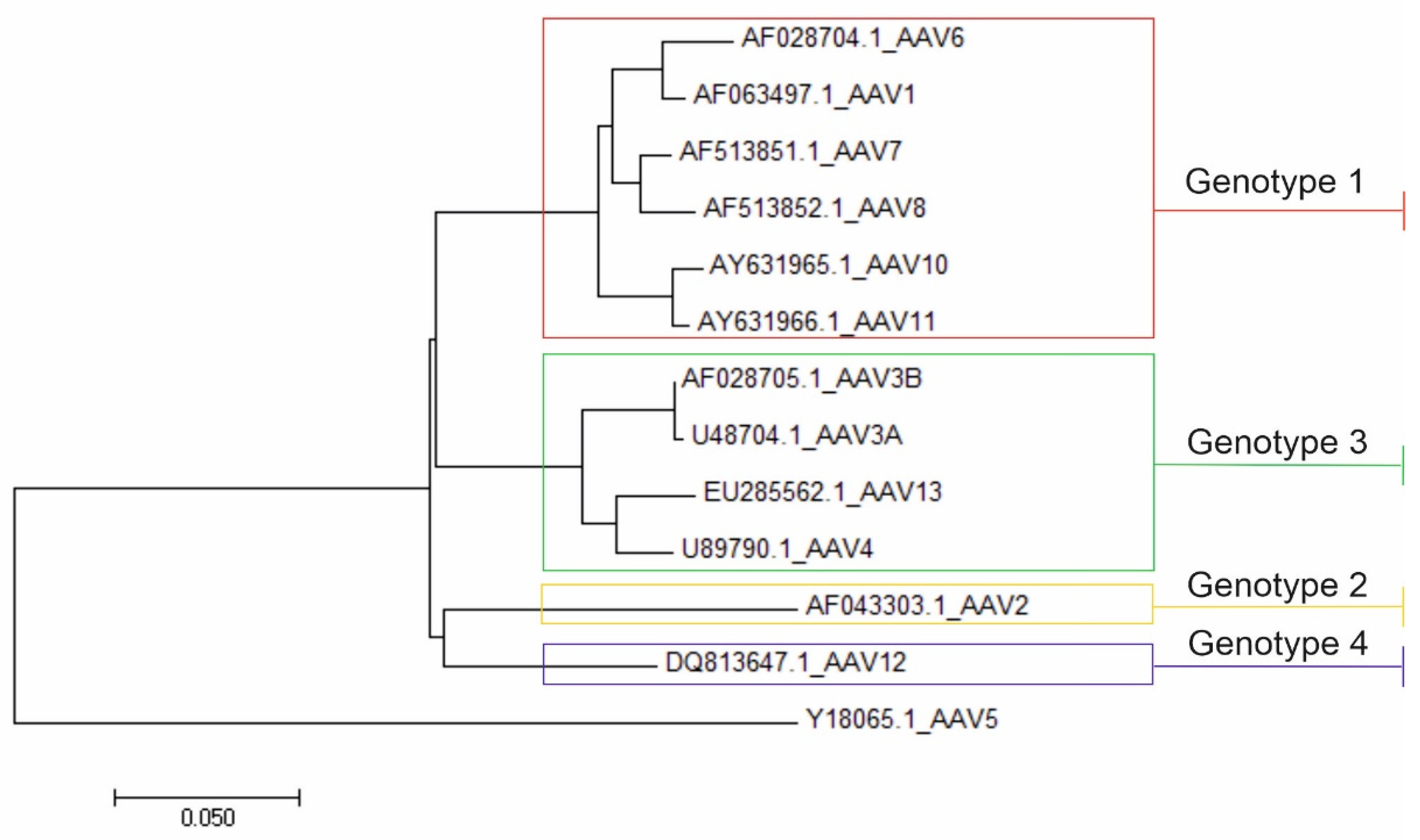
3.2. Recombination Analysis
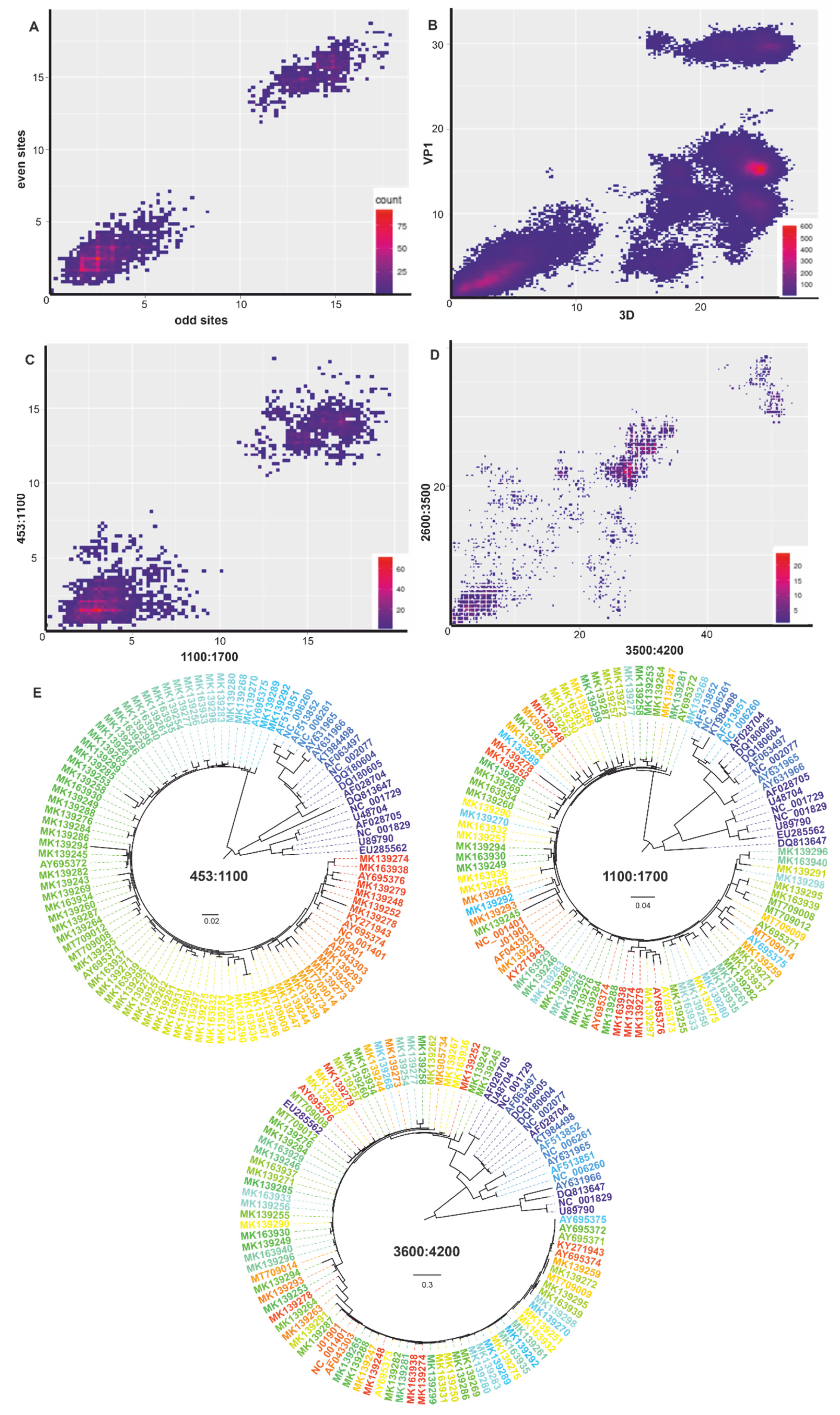
3.3. Nucleotide and Amino Acid Pairwise Distance Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Im, D.S.; Muzyczka, N. Partial purification of adeno-associated virus Rep78, Rep52, and Rep40 and their biochemical characterization. J. Virol. 1992, 66, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyöstiö, S.R.; Owens, R.A.; Weitzman, M.D.; Antoni, B.A.; Chejanovsky, N.; Carter, B.J. Analysis of adeno-associated virus (AAV) wild-type and mutant Rep proteins for their abilities to negatively regulate AAV p5 and p19 mRNA levels. J. Virol. 1994, 68, 2947–2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trempe, J.P.; Carter, B.J. Alternate mRNA splicing is required for synthesis of adeno-associated virus VP1 capsid protein. J. Virol. 1988, 62, 3356–3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, P.J.; Samulski, R.J. Adeno-associated viral vectors as gene delivery vehicles. Int. J. Mol. Med. 2000, 6, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, M.A.F.V. Adeno-associated virus: From defective virus to effective vector. Virol. J. 2005, 2, 43. [Google Scholar] [CrossRef] [Green Version]
- Bohenzky, R.A.; Lefebvre, R.B.; Berns, K.I. Sequence and symmetry requirements within the internal palindromic sequences of the adeno-associated virus terminal repeat. Virology 1988, 166, 316–327. [Google Scholar] [CrossRef]
- Wang, X.-S.; Ponnazhagan, S.; Srivastava, A. Rescue and Replication Signals of the Adeno-associated Virus 2 Genome. J. Mol. Biol. 1995, 250, 573–580. [Google Scholar] [CrossRef]
- Meier, A.F.; Fraefel, C.; Seyffert, M. The Interplay between Adeno-Associated Virus and Its Helper Viruses. Viruses 2020, 12, 662. [Google Scholar] [CrossRef]
- Parks, W.P.; Boucher, D.W.; Melnick, J.L.; Taber, L.H.; Yow, M.D. Seroepidemiological and Ecological Studies of the Adenovirus-Associated Satellite Viruses. Infect. Immun. 1970, 2, 716–722. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Vandenberghe, L.; Wilson, J. New Recombinant Serotypes of AAV Vectors. Curr. Gene Ther. 2005, 5, 285–297. [Google Scholar] [CrossRef]
- Bossis, I.; Chiorini, J.A. Cloning of an Avian Adeno-Associated Virus (AAAV) and Generation of Recombinant AAAV Particles. J. Virol. 2003, 77, 6799–6810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Katano, H.; Bossis, I.; Chiorini, J.A. Cloning and Characterization of a Bovine Adeno-Associated Virus. J. Virol. 2004, 78, 6509–6516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbetman, A.E.; Lochrie, M.; Zhou, S.; Wellman, J.; Scallan, C.; Doroudchi, M.M.; Randlev, B.; Patarroyo-White, S.; Liu, T.; Smith, P.; et al. Novel Caprine Adeno-Associated Virus (AAV) Capsid (AAV-Go.1) Is Closely Related to the Primate AAV-5 and Has Unique Tropism and Neutralization Properties. J. Virol. 2005, 79, 15238–15245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.F. Transient transfection methods for clinical adeno-associated viral vector production. Hum. Gene Ther. 2009, 20, 698–706. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Schaffer, D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014, 15, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Surosky, R.T.; Urabe, M.; Godwin, S.G.; McQuiston, S.A.; Kurtzman, G.J.; Ozawa, K.; Natsoulis, G. Adeno-associated virus Rep proteins target DNA sequences to a unique locus in the human genome. J. Virol. 1997, 71, 7951–7959. [Google Scholar] [CrossRef] [Green Version]
- Lukashev, A.N.; Zamyatnin, A.A. Viral vectors for gene therapy: Current state and clinical perspectives. Biochem. 2016, 81, 700–708. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Daya, S.; Berns, K.I. Gene Therapy Using Adeno-Associated Virus Vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago-Ortiz, J.L.; Schaffer, D.V. Adeno-associated virus (AAV) vectors in cancer gene therapy. J. Control. Release 2016, 240, 287–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nienhuis, A.W.; Nathwani, A.C.; Davidoff, A.M. Gene Therapy for Hemophilia. Mol. Ther. 2017, 25, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Ylä-Herttuala, S. Endgame: Glybera finally recommended for approval as the first gene therapy drug in the European union. Mol. Ther. 2012, 20, 1831–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spark Therapeutics LUXTURNA (voretigene neparvovec-rzyl) [Package Insert]. Available online: https://www.fda.gov/media/109906/download (accessed on 25 June 2021).
- Zhu, H.; Wang, T.; John Lye, R.; French, B.A.; Annex, B.H. Neuraminidase-mediated desialylation augments AAV9-mediated gene expression in skeletal muscle. J. Gene Med. 2018, 20, e3049. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benk, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- Hoggan, M.D.; Blacklow, N.R.; Rowe, W.P. Studies of small DNA viruses found in various adenovirus preparations: Physical, biological, and immunological characteristics. Proc. Natl. Acad. Sci. USA 1966, 55, 1467–1474. [Google Scholar] [CrossRef] [Green Version]
- Parks, W.P.; Melnick, J.L.; Rongey, R.; Mayor, H.D. Physical Assay and Growth Cycle Studies of a Defective Adeno-Satellite Virus. J. Virol. 1967, 1, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Bantel-Schaal, U.; Zur Hausen, H. Characterization of the DNA of a defective human parvovirus isolated from a genital site. Virology 1984, 134, 52–63. [Google Scholar] [CrossRef]
- Bantel-Schaal, U.; Delius, H.; Schmidt, R.; zur Hausen, H. Human Adeno-Associated Virus Type 5 Is Only Distantly Related to Other Known Primate Helper-Dependent Parvoviruses. J. Virol. 1999, 73, 939–947. [Google Scholar] [CrossRef] [Green Version]
- Murphy, F.A.; Fauquet, C.M.; Bishop, D.H.L.; Ghabrial, S.A.; Jarvis, S.A.; Martelli, G.; Mayo, P.M.A.; Summers, M.D. Virus Taxonomy. Sixth report of the International Committee on Taxonomy of Viruses. Arch. Virol. Suppl. 1995, 10, 175. [Google Scholar]
- Rutledge, E.A.; Halbert, C.L.; Russell, D.W. Infectious Clones and Vectors Derived from Adeno-Associated Virus (AAV) Serotypes Other Than AAV Type 2. J. Virol. 1998, 72, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.-P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Vandenberghe, L.H.; Alvira, M.R.; Lu, Y.; Calcedo, R.; Zhou, X.; Wilson, J.M. Clades of Adeno-Associated Viruses Are Widely Disseminated in Human Tissues. J. Virol. 2004, 78, 6381–6388. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Wang, L.; Takeuchi, T.; Kanda, T. Two novel adeno-associated viruses from cynomolgus monkey: Pseudotyping characterization of capsid protein. Virology 2004, 330, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Voutetakis, A.; Afione, S.; Zheng, C.; Mandikian, D.; Chiorini, J.A. Adeno-Associated Virus Type 12 (AAV12): A Novel AAV Serotype with Sialic Acid- and Heparan Sulfate Proteoglycan-Independent Transduction Activity. J. Virol. 2008, 82, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Govindasamy, L.; Afione, S.; Kaludov, N.; Agbandje-McKenna, M.; Chiorini, J.A. Molecular Characterization of the Heparin-Dependent Transduction Domain on the Capsid of a Novel Adeno-Associated Virus Isolate, AAV(VR-942). J. Virol. 2008, 82, 8911–8916. [Google Scholar] [CrossRef] [Green Version]
- Ronzitti, G.; Gross, D.A.; Mingozzi, F. Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front. Immunol. 2020, 11, 670. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2018, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakulenko, Y.; Deviatkin, A.; Drexler, J.F.; Lukashev, A. Modular evolution of coronavirus genomes. Viruses 2021, 13, 1270. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashev, A.N.; Elena, Y.; Belalov, I.S.; Ivanova, O.E.; Eremeeva, T.P.; Reznik, V.I.; Trotsenko, O.E.; Drexler, J.F.; Drosten, C. Recombination strategies and evolutionary dynamics of the Human enterovirus A global gene pool. J. Gen. Virol. 2014, 95, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Bao, Y.; Bavari, S.; Becker, S.; Bradfute, S.; Brister, J.R.; Bukreyev, A.A.; Chandran, K.; Davey, R.A.; Dolnik, O.; et al. Virus nomenclature below the species level: A standardized nomenclature for natural variants of viruses assigned to the family Filoviridae. Arch. Virol. 2013, 158, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Ellis, B.L.; Hirsch, M.L.; Barker, J.C.; Connelly, J.P.; Steininger, R.J.; Porteus, M.H. A survey of ex vivo/in vitro transduction efficiency of mammalian primary cells and cell lines with Nine natural adeno-associated virus (AAV1-9) and one engineered adeno-associated virus serotype. Virol. J. 2013, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A. In vivo tissue-tropism of adeno-associated viral vectors. Curr. Opin. Virol. 2016, 21, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Korneyenkov, M.A.; Zamyatnin, A.A. Next step in gene delivery: Modern approaches and further perspectives of aav tropism modification. Pharmaceutics 2021, 13, 750. [Google Scholar] [CrossRef]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Simmonds, P. Recombination and selection in the evolution of picornaviruses and other Mammalian positive-stranded RNA viruses. J. Virol. 2006, 80, 11124–11140. [Google Scholar] [CrossRef] [Green Version]
- Lukashov, V.V.; Goudsmit, J. Evolutionary Relationships among Parvoviruses: Virus-Host Coevolution among Autonomous Primate Parvoviruses and Links between Adeno-Associated and Avian Parvoviruses. J. Virol. 2001, 75, 2729–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashev, A.N. Recombination among picornaviruses. Rev. Med. Virol. 2010, 20, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Shackelton, L.A.; Hoelzer, K.; Parrish, C.R.; Holmes, E.C. Comparative analysis reveals frequent recombination in the parvoviruses. J. Gen. Virol. 2007, 88, 3294–3301. [Google Scholar] [CrossRef] [PubMed]
- Hoelzer, K.; Parrish, C.R. Evolution and Variation of the Parvoviruses, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 9780123741530. [Google Scholar]
- Martin, D.P.; Biagini, P.; Lefeuvre, P.; Golden, M.; Roumagnac, P.; Varsani, A. Recombination in eukaryotic single stranded DNA viruses. Viruses 2011, 3, 1699–1738. [Google Scholar] [CrossRef]
- Gao, G.; Alvira, M.R.; Somanathan, S.; Lu, Y.; Vandenberghe, L.H.; Rux, J.J.; Calcedo, R.; Sanmiguel, J.; Abbas, Z.; Wilson, J.M. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc. Natl. Acad. Sci. USA 2003, 100, 6081–6086. [Google Scholar] [CrossRef] [Green Version]
- Lukashev, A.N.; Ivanova, O.E.; Eremeeva, T.P.; Iggo, R.D. Evidence of frequent recombination among human adenoviruses. J. Gen. Virol. 2008, 89, 380–388. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Holmes, E.C. Phylogenetic Evidence for the Rapid Evolution of Human B19 Erythrovirus. J. Virol. 2006, 80, 3666–3669. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, M.; Ohshima, T.; Une, Y.; Yachi, A. Recombination between vaccine and field strains of canine parvovirus is revealed by isolation of virus in canine and feline cell cultures. J. Vet. Med. Sci. 2008, 70, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.; Chen, J.; Xu, Z.; Yu, J.; Huang, C.; Jin, M.; Li, H.; Zhang, M.; Jin, Y.; Duan, Z. jun Phylogenetic and recombination analysis of human bocavirus 2. BMC Infect. Dis. 2011, 11, 50. [Google Scholar] [CrossRef] [Green Version]
- Tyumentsev, A.I.; Tikunova, N.V.; Tikunov, A.Y.; Babkin, I.V. Recombination in the evolution of human bocavirus. Infect. Genet. Evol. 2014, 28, 11–14. [Google Scholar] [CrossRef]
- Xiao, W.; Chirmule, N.; Berta, S.C.; Gao, G.; Wilson, J.M.; Cullough, B.M.C. Gene Therapy Vectors Based on Adeno-Associated Virus Type 1 Gene Therapy Vectors Based on Adeno-Associated Virus Type 1. J. Virol. Methods 1999, 73, 3994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashev, A.N. Role of recombination in evolution of enteroviruses. Rev. Med. Virol. 2005, 15, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Seto, D.; Chodosh, J.; Brister, J.R.; Jones, M.S. Using the Whole-Genome Sequence To Characterize and Name Human Adenoviruses. J. Virol. 2011, 85, 5701–5702. [Google Scholar] [CrossRef] [Green Version]
- Simsek, C.; Corman, V.M.; Everling, H.U.; Lukashev, A.N.; Rasche, A.; Maganga, G.D.; Binger, T.; Jansen, D.; Beller, L.; Deboutte, W.; et al. At Least Seven Distinct Rotavirus Genotype Constellations in Bats with Evidence of Reassortment and Zoonotic Transmissions. mBio 2021, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AAV1 | AAV2 | AAV3 | AAV4 | AAV6 | AAV7 | AAV8 | AAV10 | AAV11 | AAV12 | AAV13 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AAV1 | 0 | 0.16 | 0.12 | 0.11 | 0.02 | 0.03 | 0.04 | 0.05 | 0.05 | 0.12 | 0.12 |
| AAV2 | 0.17 | 0 | 0.14 | 0.14 | 0.15 | 0.15 | 0.16 | 0.16 | 0.15 | 0.14 | 0.14 |
| AAV3 | 0.16 | 0.18 | 0 | 0.05 | 0.13 | 0.12 | 0.13 | 0.12 | 0.12 | 0.13 | 0.05 |
| AAV4 | 0.14 | 0.19 | 0.06 | 0 | 0.12 | 0.12 | 0.13 | 0.12 | 0.12 | 0.13 | 0.03 |
| AAV6 | 0.02 | 0.18 | 0.16 | 0.15 | 0 | 0.05 | 0.06 | 0.07 | 0.06 | 0.13 | 0.13 |
| AAV7 | 0.03 | 0.16 | 0.16 | 0.15 | 0.04 | 0 | 0.03 | 0.05 | 0.04 | 0.12 | 0.12 |
| AAV8 | 0.05 | 0.18 | 0.18 | 0.16 | 0.05 | 0.03 | 0 | 0.05 | 0.05 | 0.13 | 0.14 |
| AAV10 | 0.07 | 0.17 | 0.16 | 0.16 | 0.08 | 0.07 | 0.07 | 0 | 0.01 | 0.13 | 0.13 |
| AAV11 | 0.07 | 0.16 | 0.16 | 0.15 | 0.07 | 0.06 | 0.06 | 0.02 | 0 | 0.12 | 0.13 |
| AAV12 | 0.15 | 0.19 | 0.19 | 0.17 | 0.15 | 0.14 | 0.16 | 0.16 | 0.16 | 0 | 0.13 |
| AAV13 | 0.15 | 0.18 | 0.06 | 0.02 | 0.15 | 0.15 | 0.16 | 0.16 | 0.15 | 0.18 | 0 |
| Genotype 1 | Genotype 2 | Genotype 3 | Genotype 4 | |
|---|---|---|---|---|
| Genotype 1 (AAV1 + AAV6 + AAV7 + AAV8 + AAV10 + AAV11) | 0 | 0.15–0.16 | 0.11–0.14 | 0.12–0.13 |
| Genotype 2 (AAV 2) | - | 0 | 0.14–0.15 | 0.14 |
| Genotype 3 (AAV3 + AAV4 + AAV13) | - | - | 0 | 0.13 |
| Genotype 4 (AVV12) | - | - | - | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beloukhova, M.I.; Lukashev, A.N.; Volchkov, P.Y.; Zamyatnin, A.A., Jr.; Deviatkin, A.A. Robust AAV Genotyping Based on Genetic Distances in Rep Gene That Are Maintained by Ubiquitous Recombination. Viruses 2022, 14, 1038. https://doi.org/10.3390/v14051038
Beloukhova MI, Lukashev AN, Volchkov PY, Zamyatnin AA Jr., Deviatkin AA. Robust AAV Genotyping Based on Genetic Distances in Rep Gene That Are Maintained by Ubiquitous Recombination. Viruses. 2022; 14(5):1038. https://doi.org/10.3390/v14051038
Chicago/Turabian StyleBeloukhova, Marina I., Alexander N. Lukashev, Pavel Y. Volchkov, Andrey A. Zamyatnin, Jr., and Andrei A. Deviatkin. 2022. "Robust AAV Genotyping Based on Genetic Distances in Rep Gene That Are Maintained by Ubiquitous Recombination" Viruses 14, no. 5: 1038. https://doi.org/10.3390/v14051038
APA StyleBeloukhova, M. I., Lukashev, A. N., Volchkov, P. Y., Zamyatnin, A. A., Jr., & Deviatkin, A. A. (2022). Robust AAV Genotyping Based on Genetic Distances in Rep Gene That Are Maintained by Ubiquitous Recombination. Viruses, 14(5), 1038. https://doi.org/10.3390/v14051038