
Clinical, Laboratory, and Molecular Epidemiology of an Outbreak of Aseptic Meningitis Due to a Triple-Recombinant Echovirus in Ashburton, New Zealand

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
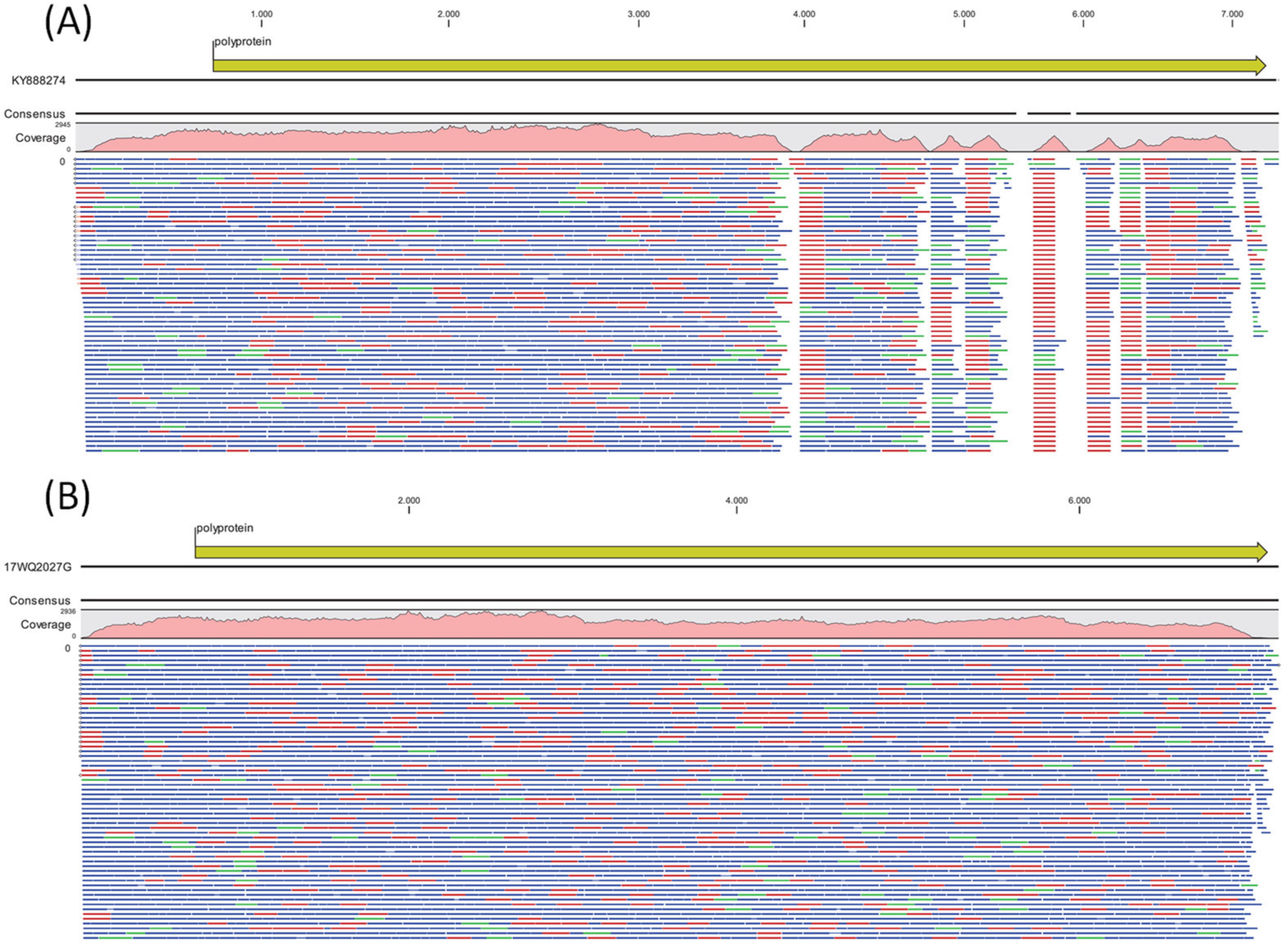
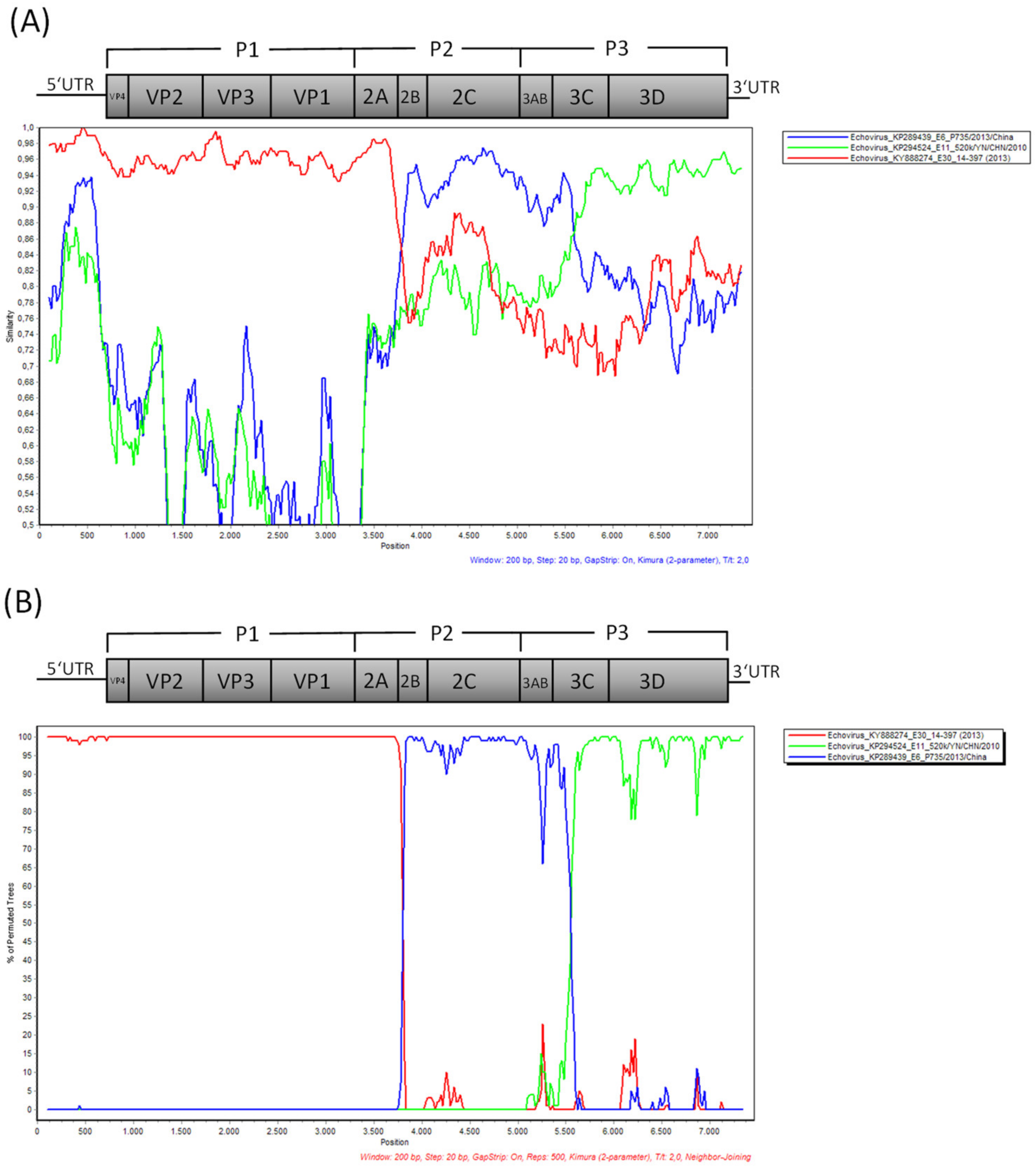
3. Results
3.1. Clinical Findings
3.2. Laboratory Findings
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Melnick, J.L. Enteroviruses: Polioviruses, Coxsackiviruses, Echoviruses, and Newer Enteroviruses. In Fields Virology, 3rd ed.; Knipe, D.M.F.B.H.P., Ed.; Lippincott-Raven: Philadelphia, PA, USA, 1996; pp. 655–712. [Google Scholar]
- Lema, C.; Torres, C.; Van der Sanden, S. Gobal phylodynamics of Echovirus 30 revealed differential behaviour among viral lineages. Virology 2019, 531, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Ye, H.; Yanjun, Z.; Xie, G.; Cui, D.; Yu, C.; Wang, Y.; Yang, X.; Zhou, F.; Zhang, Y.; et al. Laboratory diagnosis and genetic analysis of a family clustering outbreak of aseptic meningitis due to echovirus 30. Pathog. Glob. Health 2016, 110, 233. [Google Scholar] [CrossRef] [PubMed]
- Croker, C.; Civen, R.; Keough, K.; Ngo, V.; Marutani, A.; Schwartz, B. Aseptic meningitis outbreak associated with echovirus 30 among high school football players—Los Angeles County, California 2014. MMWR Morb. Mortal Wkly. Rep. 2015, 63, 1228. [Google Scholar] [PubMed]
- Bennett, S.; Harvala, H.; Witteveldt, J.; Leitch, E.C.M.; McLeish, N.; Templeton, K.; Gunson, R.; Carman, W.F.; Simmonds, P. Rapid simultaneous detection of enterovirus and parechovirus RNAs in clinical samples by one-step real-time reverse trasncription-PCR assay. J. Clin. Microbiol. 2011, 49, 2620–2624. [Google Scholar] [CrossRef] [PubMed]
- Nix, W.A.; Oberste, M.S.; Pallansch, M.A. Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J. Clin. Microbiol. 2006, 44, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.; Dvorkin, M.; Kulikov, A.; Lesin, V.M.; Nikolenko, S.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Price, M.; Dehal, P.; Arkin, A.P.A. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, M.; Goto, A.; Komagome, R.; Yamaguchi, H.; Maruo, Y.; Nakanishi, M.; Ishida, S.; Nagano, H.; Sugisawa, T.; Okano, M. Genetic characteriziation of a novel recombinant echovirus 30 strain causing a regional epidemic of aseptic meningitis in Hokkaido, Japan, 2017. Arch. Virol. 2020, 165, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Gámbaro, F.; Pérez, A.B.; Agüera, E.; Prot, M.; Martínez-Martínez, L.; Cabrerizo, M.; Simon-Loriere, E.; Fernandez-Garcia, M.D. Genomic surveillance of enterovirus associated with aseptic meningitis cases in southern Spain, 2015–2018. Sci. Rep. 2021, 11, 21523. [Google Scholar] [CrossRef] [PubMed]
- Cernescu, C.; Târdei, G.; Ruta, S.; Bleotu, C.; Alexiu, I.; Jucu, V. An outbreak of aseptic meningitis due to ECHO 30 virus in Romania during the 1999 summer. Rom. J. Virol. 1999, 50, 99–106. [Google Scholar]
- Oberste, M.S.; Maher, K.; Kennett, M.L.; Campbell, J.J.; Carpenter, M.S.; Schnurr, D.; Pallansch, M.A. Molecular Epidemiology and Genetic Diversity of Echovirus Type 30 (E30): Genotypes Correlate with Temporal Dynamics of E30 Isolation. J. Clin. Microbiol. 1999, 37, 3928–3933. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Huang, K.; Li, L.; Wu, X.; Zheng, L.; Wan, C.; Zhao, W.; Ke, C.; Zhang, B. Complete genome sequence analysis of human echovirus 30 isolated during a large outbreak in Guangdong Province of China in 2012. Arch. Virol. 2014, 159, 379–383. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McWilliam Leitch, E.C.; Bending, J.M. Transmission networks and population turnover of echovirus 30. J. Virol. 2009, 83, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Lowry, K.; Woodman, A.; Cook, J.; Evans, D.J. Recombination in enteroviruses is a biphasic replicative process involving the generation of greater-than genome length ‘imprecise’ intermediates. PLoS Pathog. 2014, 10, e1004191. [Google Scholar] [CrossRef] [PubMed]
- Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses 2019, 11, 859. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dilcher, M.; Howard, J.C.; Dalton, S.C.; Anderson, T.; Clinghan, R.T.; Werno, A.M. Clinical, Laboratory, and Molecular Epidemiology of an Outbreak of Aseptic Meningitis Due to a Triple-Recombinant Echovirus in Ashburton, New Zealand. Viruses 2022, 14, 658. https://doi.org/10.3390/v14040658
Dilcher M, Howard JC, Dalton SC, Anderson T, Clinghan RT, Werno AM. Clinical, Laboratory, and Molecular Epidemiology of an Outbreak of Aseptic Meningitis Due to a Triple-Recombinant Echovirus in Ashburton, New Zealand. Viruses. 2022; 14(4):658. https://doi.org/10.3390/v14040658
Chicago/Turabian StyleDilcher, Meik, Julia C. Howard, Simon C. Dalton, Trevor Anderson, Richard T. Clinghan, and Anja M. Werno. 2022. "Clinical, Laboratory, and Molecular Epidemiology of an Outbreak of Aseptic Meningitis Due to a Triple-Recombinant Echovirus in Ashburton, New Zealand" Viruses 14, no. 4: 658. https://doi.org/10.3390/v14040658
APA StyleDilcher, M., Howard, J. C., Dalton, S. C., Anderson, T., Clinghan, R. T., & Werno, A. M. (2022). Clinical, Laboratory, and Molecular Epidemiology of an Outbreak of Aseptic Meningitis Due to a Triple-Recombinant Echovirus in Ashburton, New Zealand. Viruses, 14(4), 658. https://doi.org/10.3390/v14040658