
Aminoacyl-tRNA Synthetase: A Non-Negligible Molecule in RNA Viral Infection


Abstract
:1. Introduction
2. Potential Roles of aaRSs in Infections with Coronaviruses
3. Host aaRSs Are Involved in Host Innate Immune Responses to RNA Viral Infections
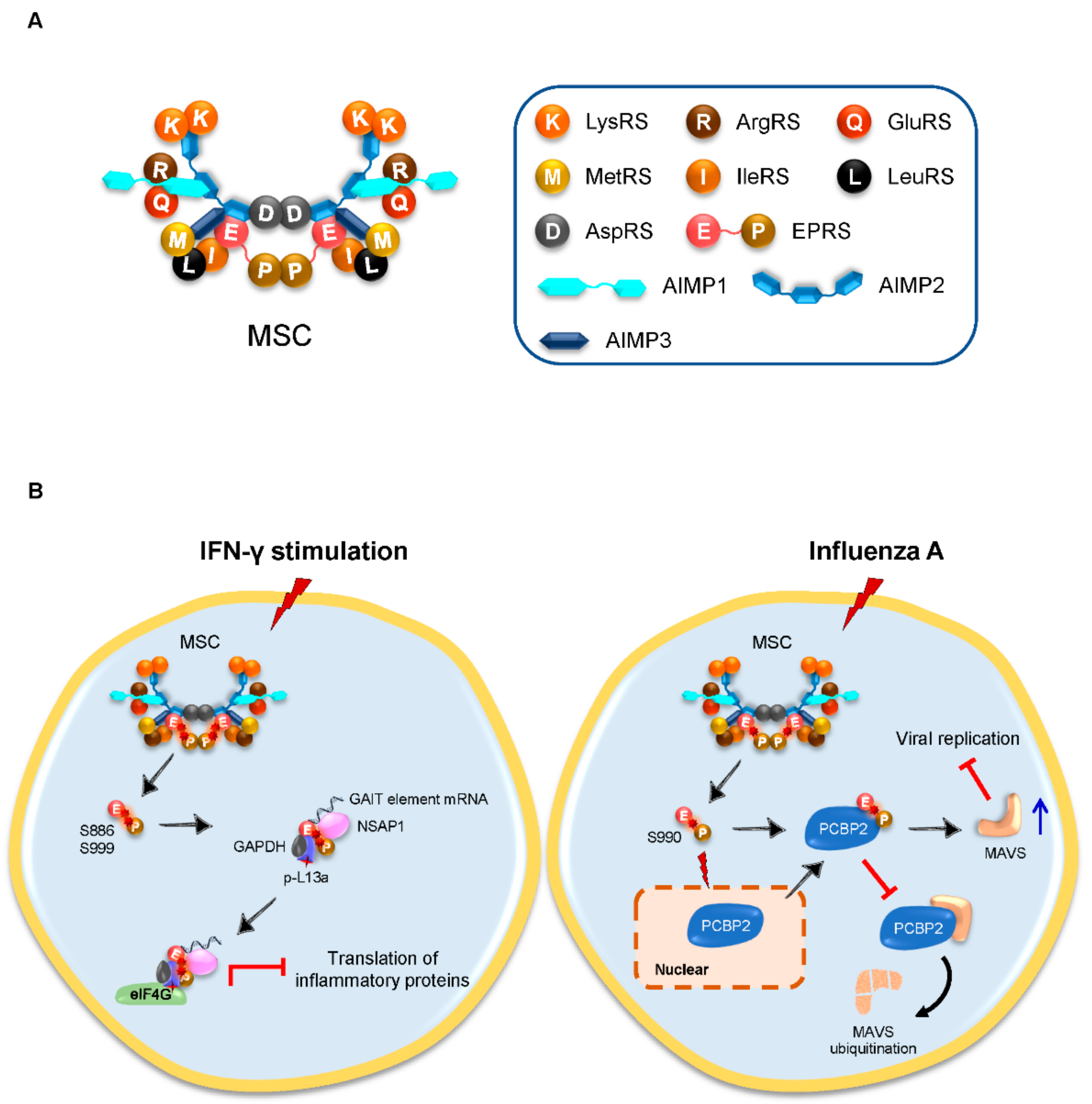
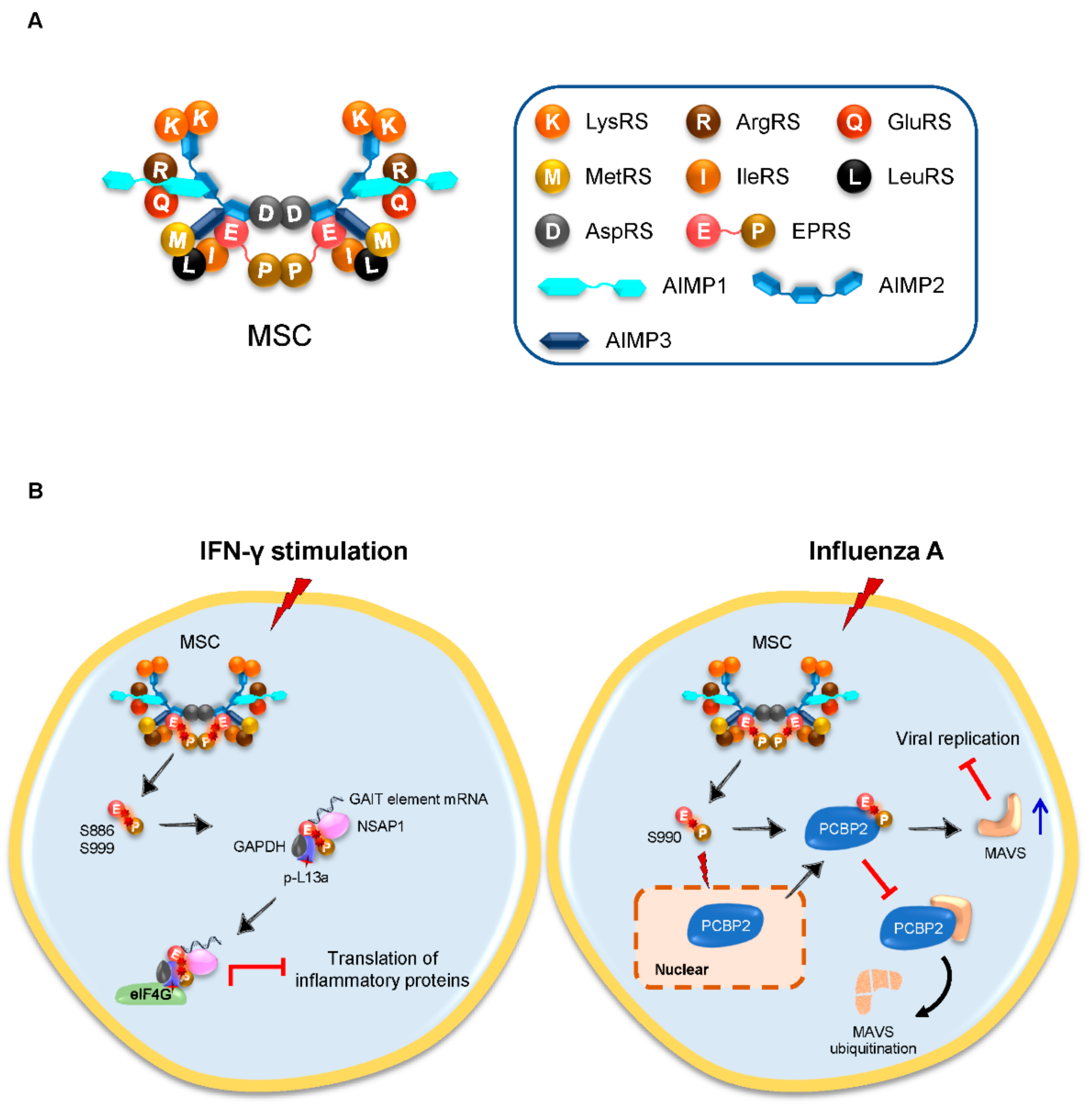
3.1. Inducible Released EPRS Regulates Innate Immune Responses
3.2. ThrRS Stimulates the Activation and Maturation of DCs
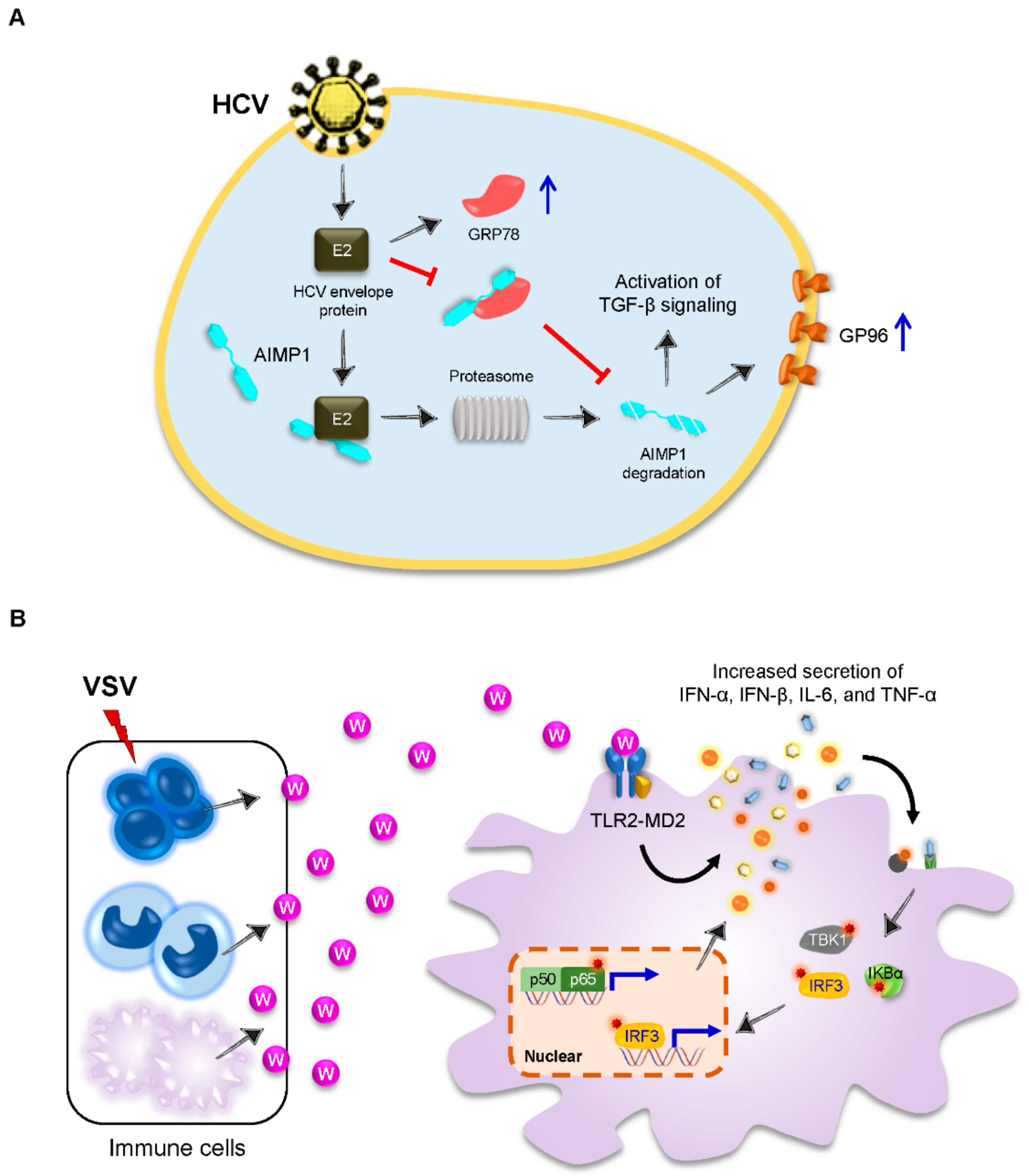
3.3. AIMP1 Is Secreted to Stimulate Innate Antiviral Immune Responses
3.4. Tryptophanyl-tRNA Synthetase (TrpRS) Elicits Innate Immune Response as a Cytokine
4. Host aaRSs Are Employed by RNA Viruses for Invasion
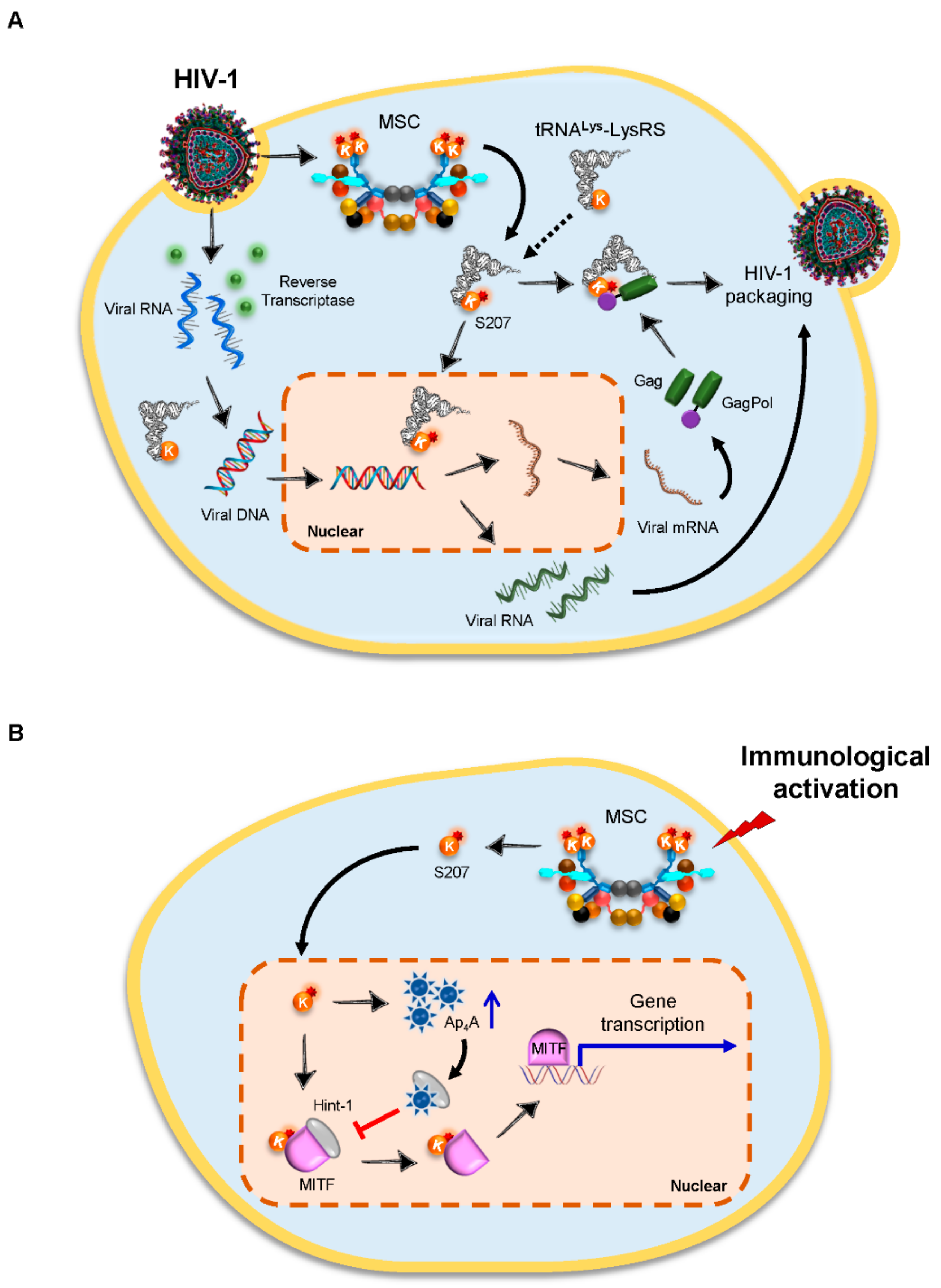
4.1. Lysyl-tRNA Synthetase (LysRS) Is an Essential Host Factor for the Packaging of HIV-1 Virions
4.2. Glycyl-tRNA Synthetase (GlyRS) Regulates Viral Translation Initiation via the IRES Element
4.3. TrpRS Functions as a Cellular Entry Factor for Enteroviruses
5. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ling, J.; Reynolds, N.; Ibba, M. Aminoacyl-tRNA synthesis and translational quality control. Annu. Rev. Microbiol. 2009, 63, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Yang, X.L.; Schimmel, P. New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 2010, 11, 668–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Schimmel, P. Essential nontranslational functions of tRNA synthetases. Nat. Chem. Biol. 2013, 9, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [PubMed]
- Feng, Y.; Tang, K.; Lai, Q.; Liang, J.; Feng, M.; Zhou, Z.W.; Cui, H.; Du, X.; Zhang, H.; Sun, L. The landscape of aminoacyl-tRNA synthetases involved in severe acute respiratory syndrome coronavirus 2 infection. Front. Physiol. 2021, 12, 818297. [Google Scholar] [CrossRef] [PubMed]
- Refolo, G.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Ciccosanti, F. Mitochondrial interactome: A focus on antiviral signaling pathways. Front. Cell Dev. Biol. 2020, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.Z.; Wang, S.Y.; Zheng, Z.Q.; Huang, Y.; Li, W.W.; Xu, Z.S.; Wang, Y.Y. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell. Mol. Immunol. 2021, 18, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, D.Y.; Kim, J.H.; Ahn, T.J.; Cho, Y.; Hwang, D.; Kim, S. Evolution of the multi-tRNA synthetase complex and its role in cancer. J. Biol. Chem. 2019, 294, 5340–5351. [Google Scholar] [CrossRef] [Green Version]
- Sampath, P.; Mazumder, B.; Seshadri, V.; Gerber, C.A.; Chavatte, L.; Kinter, M.; Ting, S.M.; Dignam, J.D.; Kim, S.; Driscoll, D.M.; et al. Noncanonical function of glutamyl-prolyl-tRNA synthetase: Gene-specific silencing of translation. Cell 2004, 119, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.S.; Arif, A.; Fox, P.L. Macromolecular complexes as depots for releasable regulatory proteins. Trends Biochem. Sci. 2007, 32, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Cho, B.H.; Park, S.G.; Kim, S. Aminoacyl-tRNA synthetase complexes: Beyond translation. J. Cell Sci. 2004, 117 Pt 17, 3725–3734. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Han, J.M.; Kim, S. Protein-protein interactions and multi-component complexes of aminoacyl-tRNA synthetases. Top Curr Chem. 2014, 344, 119–144. [Google Scholar] [PubMed]
- Cho, H.Y.; Maeng, S.J.; Cho, H.J.; Choi, Y.S.; Chung, J.M.; Lee, S.; Kim, H.K.; Kim, J.H.; Eom, C.Y.; Kim, Y.G.; et al. Assembly of multi-tRNA Synthetase complex via heterotetrameric glutathione transferase-homology domains. J. Biol. Chem. 2015, 290, 29313–29328. [Google Scholar] [CrossRef] [Green Version]
- Arif, A.; Jia, J.; Mukhopadhyay, R.; Willard, B.; Kinter, M.; Fox, P.L. Two-site phosphorylation of EPRS coordinates multimodal regulation of noncanonical translational control activity. Mol. Cell 2009, 35, 164–180. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Arif, A.; Ray, P.S.; Fox, P.L. WHEP domains direct noncanonical function of glutamyl-Prolyl tRNA synthetase in translational control of gene expression. Mol. Cell 2008, 29, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Vyas, K.; Chaudhuri, S.; Leaman, D.W.; Komar, A.A.; Musiyenko, A.; Barik, S.; Mazumder, B. Genome-wide polysome profiling reveals an inflammation-responsive posttranscriptional operon in gamma interferon-activated monocytes. Mol. Cell. Biol. 2009, 29, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Galán, C.; Sola, I.; Nogales, A.; Thomas, B.; Akoulitchev, A.; Enjuanes, L.; Almazán, F. Host cell proteins interacting with the 3’ end of TGEV coronavirus genome influence virus replication. Virology 2009, 391, 304–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquez-Jurado, S.; Nogales, A.; Zuniga, S.; Enjuanes, L.; Almazan, F. Identification of a gamma interferon-activated inhibitor of translation-like RNA motif at the 3’ end of the transmissible gastroenteritis coronavirus genome modulating innate immune response. mBio 2015, 6, e00105. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Y.; Lee, H.C.; Kim, H.K.; Jang, S.Y.; Park, S.J.; Kim, Y.H.; Kim, J.H.; Hwang, J.; Kim, J.H.; Kim, T.H.; et al. Infection-specific phosphorylation of glutamyl-prolyl tRNA synthetase induces antiviral immunity. Nat. Immunol. 2016, 17, 1252–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.J.; Park, S.H.; Cho, K.M.; Jung, K.I.; Cho, D.; Kim, T.S. Threonyl-tRNA synthetase promotes T helper type 1 cell responses by inducing dendritic cell maturation and IL-12 production via an NF-kappaB pathway. Front. Immunol. 2020, 11, 571959. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Liu, Y.; Yin, Y.; Shao, A.; Zhang, B.; Kim, S.; Zhou, J. MSC p43 required for axonal development in motor neurons. Proc. Natl. Acad. Sci. USA 2009, 106, 15944–15949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinstein, M.; Markus, B.; Noyman, I.; Shalev, H.; Flusser, H.; Shelef, I.; Liani-Leibson, K.; Shorer, Z.; Cohen, I.; Khateeb, S.; et al. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am. J. Hum. Genet. 2010, 87, 820–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Son, M.K.; Jung, K.H.; Kim, K.; Kim, G.J.; Lee, S.H.; Hong, S.S.; Park, S.G. Aminoacyl-tRNA synthetase interacting multi-functional protein 1 attenuates liver fibrosis by inhibiting TGFbeta signaling. Int. J. Oncol. 2016, 48, 747–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, C.; Gu, C.; Zhao, M.; Zhu, D.; Wang, N.; Yu, J.; Yao, Y.; Chen, Y.; Shi, M.; Gu, Q.; et al. The role of the AIMP1 pathway in diabetic retinopathy: AIMP1-targeted intervention study in diabetic retinopathy. Ophthalmic Res. 2020, 63, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Hur, S.Y.; Kim, Y.R.; Yoo, N.J.; Lee, S.H. Expression of AIMP1, 2 and 3, the scaffolds for the multi-tRNA synthetase complex, is downregulated in gastric and colorectal cancer. Tumori J. 2011, 97, 380–385. [Google Scholar] [CrossRef]
- Cheng, W.; Ren, X.; Zhang, C.; Cai, J.; Liu, Y.; Han, S.; Wu, A. Bioinformatic profiling identifies an immune-related risk signature for glioblastoma. Neurology 2016, 86, 2226–2234. [Google Scholar] [CrossRef]
- Gao, W.; An, C.; Xue, X.; Zheng, X.; Niu, M.; Zhang, Y.; Liu, H.; Zhang, C.; Lu, Y.; Cui, J.; et al. Mass spectrometric analysis identifies AIMP1 and LTA4H as FSCN1-binding proteins in laryngeal squamous cell carcinoma. Proteomics 2019, 19, e1900059. [Google Scholar] [CrossRef]
- Ko, Y.G.; Park, H.; Kim, T.; Lee, J.W.; Park, S.G.; Seol, W.; Kim, J.E.; Lee, W.H.; Kim, S.H.; Park, J.E.; et al. A cofactor of tRNA synthetase, p43, is secreted to up-regulate proinflammatory genes. J. Biol. Chem. 2001, 276, 23028–23033. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Park, S.G.; Lee, J.W.; Kim, T.; Kim, G.; Ko, Y.G.; Kim, S. Monocyte cell adhesion induced by a human aminoacyl-tRNA synthetase-associated factor, p43: Identification of the related adhesion molecules and signal pathways. J. Leukoc. Biol. 2002, 71, 223–230. [Google Scholar]
- Park, S.G.; Shin, H.; Shin, Y.K.; Lee, Y.; Choi, E.C.; Park, B.J.; Kim, S. The novel cytokine p43 stimulates dermal fibroblast proliferation and wound repair. Am. J. Pathol. 2005, 166, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Kim, S.H.; Kim, S.; Kim, T.S. The novel cytokine p43 induces IL-12 production in macrophages via NF-kappaB activation, leading to enhanced IFN-gamma production in CD4+ T cells. J. Immunol. 2006, 176, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Kim, S.H.; Kim, S.; Cho, D.; Kim, T.S. AIMP1/p43 protein induces the maturation of bone marrow-derived dendritic cells with T helper type 1-polarizing ability. J. Immunol. 2008, 180, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, T.S. Aminoacyl tRNA synthetase-interacting multifunctional protein 1 acts as a novel B cell-activating factor in vitro and in vivo. J. Immunol. 2015, 194, 4729–4736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Song, J.H.; Cohen, E.P.; Cho, D.; Kim, T.S. Aminoacyl tRNA synthetase—Interacting multifunctional protein 1 activates NK cells via macrophages in vitro and in vivo. J. Immunol. 2017, 198, 4140–4147. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Tian, L.; You, R.; Halpert, M.M.; Konduri, V.; Baig, Y.C.; Paust, S.; Kim, D.; Kim, S.; Jia, F.; et al. AIMp1 potentiates TH1 polarization and is critical for effective antitumor and antiviral immunity. Front. Immunol. 2017, 8, 1801. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, S.; Myung, H. Degradation of AIMP1/p43 induced by hepatitis C virus E2 leads to upregulation of TGF-beta signaling and increase in surface expression of gp96. PLoS ONE 2014, 9, e96302. [Google Scholar]
- Ahn, Y.H.; Park, S.; Choi, J.J.; Park, B.K.; Rhee, K.H.; Kang, E.; Ahn, S.; Lee, C.H.; Lee, J.S.; Inn, K.S.; et al. Secreted tryptophanyl-tRNA synthetase as a primary defence system against infection. Nat. Microbiol. 2016, 2, 16191. [Google Scholar] [CrossRef]
- Lee, H.C.; Lee, E.S.; Uddin, M.B.; Kim, T.H.; Kim, J.H.; Chathuranga, K.; Chathuranga, W.G.; Jin, M.; Kim, S.; Kim, C.J.; et al. Released tryptophanyl-tRNA synthetase stimulates innate immune responses against viral infection. J. Virol. 2019, 93, e01291-18. [Google Scholar] [CrossRef] [Green Version]
- Javanbakht, H.; Cen, S.; Musier-Forsyth, K.; Kleiman, L. Correlation between tRNALys3 aminoacylation and its incorporation into HIV-1. J. Biol. Chem. 2002, 277, 17389–17396. [Google Scholar] [CrossRef] [Green Version]
- Cen, S.; Khorchid, A.; Javanbakht, H.; Gabor, J.; Stello, T.; Shiba, K.; Musier-Forsyth, K.; Kleiman, L. Incorporation of lysyl-tRNA synthetase into human immunodeficiency virus type 1. J. Virol. 2001, 75, 5043–5048. [Google Scholar] [CrossRef] [Green Version]
- Gabor, J.; Cen, S.; Javanbakht, H.; Niu, M.; Kleiman, L. Effect of altering the tRNA(Lys)(3) concentration in human immunodeficiency virus type 1 upon its annealing to viral RNA, GagPol incorporation, and viral infectivity. J. Virol. 2002, 76, 9096–9102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Cen, S.; Niu, M.; Javanbakht, H.; Kleiman, L. Specific inhibition of the synthesis of human lysyl-tRNA synthetase results in decreases in tRNA(Lys) incorporation, tRNA(3)(Lys) annealing to viral RNA, and viral infectivity in human immunodeficiency virus type 1. J. Virol. 2003, 77, 9817–9822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cen, S.; Javanbakht, H.; Niu, M.; Kleiman, L. Ability of wild-type and mutant lysyl-tRNA synthetase to facilitate tRNA(Lys) incorporation into human immunodeficiency virus type 1. J. Virol. 2004, 78, 1595–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewan, V.; Wei, M.; Kleiman, L.; Musier-Forsyth, K. Dual role for motif 1 residues of human lysyl-tRNA synthetase in dimerization and packaging into HIV-1. J. Biol. Chem. 2012, 287, 41955–41962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchon, A.A.; St Gelais, C.; Titkemeier, N.; Hatterschide, J.; Wu, L.; Musier-Forsyth, K. HIV-1 Exploits a dynamic multi-aminoacyl-tRNA synthetase complex to enhance viral replication. J. Virol. 2017, 91, e01240-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolkunova, E.; Park, H.; Xia, J.; King, M.P.; Davidson, E. The human lysyl-tRNA synthetase gene encodes both the cytoplasmic and mitochondrial enzymes by means of an unusual alternative splicing of the primary transcript. J. Biol. Chem. 2000, 275, 35063–35069. [Google Scholar] [CrossRef] [Green Version]
- Kaminska, M.; Shalak, V.; Francin, M.; Mirande, M. Viral hijacking of mitochondrial lysyl-tRNA synthetase. J. Virol. 2007, 81, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Kobbi, L.; Octobre, G.; Dias, J.; Comisso, M.; Mirande, M. Association of mitochondrial lysyl-tRNA synthetase with HIV-1 Gagpol involves catalytic domain of the synthetase and transframe and integrase domains of Pol. J. Mol. Biol. 2011, 410, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Kobbi, L.; Dias, J.; Comisso, M.; Mirande, M. Association of human mitochondrial lysyl-tRNA synthetase with HIV-1 GagPol does not require other viral proteins. Biochim. Open 2016, 2, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Khoder-Agha, F.; Dias, J.M.; Comisso, M.; Mirande, M. Characterization of association of human mitochondrial lysyl-tRNA synthetase with HIV-1 Pol and tRNA3(Lys). BMC Biochem. 2018, 19, 2. [Google Scholar] [CrossRef] [Green Version]
- Kovaleski, B.J.; Kennedy, R.; Hong, M.K.; Datta, S.A.; Kleiman, L.; Rein, A.; Musier-Forsyth, K. In vitro characterization of the interaction between HIV-1 Gag and human lysyl-tRNA synthetase. J. Biol. Chem. 2006, 281, 19449–19456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.P.; Saadatmand, J.; Kleiman, L.; Musier-Forsyth, K. Molecular mimicry of human tRNALys anti-codon domain by HIV-1 RNA genome facilitates tRNA primer annealing. RNA 2013, 19, 219–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Comandur, R.; Jones, C.P.; Tsang, P.; Musier-Forsyth, K. Anticodon-like binding of the HIV-1 tRNA-like element to human lysyl-tRNA synthetase. RNA 2016, 22, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Comandur, R.; Olson, E.D.; Musier-Forsyth, K. Conservation of tRNA mimicry in the 5’-untranslated region of distinct HIV-1 subtypes. RNA 2017, 23, 1850–1859. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.N.; Nechushtan, H.; Figov, N.; Razin, E. The function of lysyl-tRNA synthetase and Ap4A as signaling regulators of MITF activity in FcepsilonRI-activated mast cells. Immunity 2004, 20, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Carmi-Levy, I.; Motzik, A.; Ofir-Birin, Y.; Yagil, Z.; Yang, C.M.; Kemeny, D.M.; Han, J.M.; Kim, S.; Kay, G.; Nechushtan, H.; et al. Importin beta plays an essential role in the regulation of the LysRS-Ap(4)A pathway in immunologically activated mast cells. Mol. Cell. Biol. 2011, 31, 2111–2121. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Ma, L.; Song, C.; Xing, H.; Cen, S.; Lin, W. Anti-HIV effects of baculiferins are regulated by the potential target protein DARS. ACS Chem. Biol. 2021, 16, 1377–1389. [Google Scholar] [CrossRef]
- Belsham, G.J. Divergent picornavirus IRES elements. Virus Res. 2009, 139, 183–192. [Google Scholar] [CrossRef]
- Andreev, D.E.; Hirnet, J.; Terenin, I.M.; Dmitriev, S.E.; Niepmann, M.; Shatsky, I.N. Glycyl-tRNA synthetase specifically binds to the poliovirus IRES to activate translation initiation. Nucleic Acids Res. 2012, 40, 5602–5614. [Google Scholar] [CrossRef] [Green Version]
- Nangle, L.A.; Zhang, W.; Xie, W.; Yang, X.L.; Schimmel, P. Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc. Natl. Acad. Sci. USA 2007, 104, 11239–11244. [Google Scholar] [CrossRef] [Green Version]
- de Breyne, S.; Yu, Y.; Unbehaun, A.; Pestova, T.V.; Hellen, C.U. Direct functional interaction of initiation factor eIF4G with type 1 internal ribosomal entry sites. Proc. Natl. Acad. Sci. USA 2009, 106, 9197–9202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, M.L.; Jia, L.; Yip, C.C.; Chan, J.F.; Teng, J.L.; Chan, K.H.; Cai, J.P.; Zhang, C.; Zhang, A.J.; Wong, W.M.; et al. Human tryptophanyl-tRNA synthetase is an IFN-gamma-inducible entry factor for Enterovirus. J. Clin. Investig. 2018, 128, 5163–5177. [Google Scholar] [CrossRef] [PubMed]
- Ochsner, U.A.; Sun, X.; Jarvis, T.; Critchley, I.; Janjic, N. Aminoacyl-tRNA synthetases: Essential and still promising targets for new anti-infective agents. Expert Opin. Investig. Drugs 2007, 16, 573–593. [Google Scholar] [CrossRef] [PubMed]
- Francklyn, C.S.; Mullen, P. Progress and challenges in aminoacyl-tRNA synthetase-based therapeutics. J. Biol. Chem. 2019, 294, 5365–5385. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.; Jiang, A.; Fikrig, E. A potent prolyl tRNA synthetase inhibitor antagonizes Chikungunya and Dengue viruses. Antivir. Res. 2019, 161, 163–168. [Google Scholar] [CrossRef]
- Clarke, P.; Leser, J.S.; Bowen, R.A.; Tyler, K.L. Virus-induced transcriptional changes in the brain include the differential expression of genes associated with interferon, apoptosis, interleukin 17 receptor A, and glutamate signaling as well as flavivirus-specific upregulation of tRNA synthetases. mBio 2014, 5, e00902-14. [Google Scholar] [CrossRef] [Green Version]
- Pastorino, B.; Boucomont-Chapeaublanc, E.; Peyrefitte, C.N.; Belghazi, M.; Fusaï, T.; Rogier, C.; Tolou, H.J.; Almeras, L. Identification of cellular proteome modifications in response to West Nile virus infection. Mol. Cell. Proteom. 2009, 8, 1623–1637. [Google Scholar] [CrossRef] [Green Version]
- Colussi, T.M.; Costantino, D.A.; Hammond, J.A.; Ruehle, G.M.; Nix, J.C.; Kieft, J.S. The structural basis of transfer RNA mimicry and conformational plasticity by a viral RNA. Nature 2014, 511, 366–369. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| aaRS | RNA Virus | Effect | Interactor | Mechanisms | Ref. | |
|---|---|---|---|---|---|---|
| Regulate innate immune responses | mt-ThrRS | SARS-CoV-2 | Antivirus | SARS-CoV-2 M protein | - | [5] |
| EPRS, ArgRS | TGEV | Inhibit innate immune responses | GAIT-like RNA motif of TGEV genome | Binding to GAIT-like RNA motif at 3′-end of the TGEV genome to counteract the host innate immune response | [18] | |
| EPRS | H1N1 influenza A; VSV | Antivirus | PCBP2 | Stabilizing MAVS from PCBP2-mediated ubiquitination to facilitate antiviral function | [19] | |
| ThrRS | H1N1 influenza A | Antivirus | - | Inducing DC maturation and IL-12 production via promoting Th1 responses | [20] | |
| AIMP1 | H3N2 influenza A | Antivirus | - | Activating MAPK signaling and Th1 polarization in LPS-treated BMDCs | [35] | |
| AIMP1 | HCV | - | HCV envelope protein E2 | Inducing AIMP1 degradation to enhance TGF-β signaling and cell surface expression of GP96 | [36] | |
| TrpRS | VSV | Antivirus | TLR4-MD2 | Inducing proinflammatory cytokines and type I IFNs | [37,38] | |
| Facilitate viral infection | LysRS | HIV-1 | Virus packaging | Gag, GagPol | Activating MEK pathway; translocating to the nucleus | [45] |
| LysRS | HIV-1 | Annealing of tRNA to viral RNA | TLE of HIV-1 genome | Binding to TLE near the primer-binding site within the HIV-1 genome to increase the efficiency of tRNALys3 primer annealing | [52,53] | |
| GlyRS | PV | RNA translation initiation | PV IRES | Binding to the domain V of the PV IRES which mimics the anticodon stem-loop of tRNAGly | [59] | |
| TrpRS | EV-A71 | Virus entry | EV-A71 | Binding to EV-A71 viral proteins to enhance viral internalization | [62] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, M.; Zhang, H. Aminoacyl-tRNA Synthetase: A Non-Negligible Molecule in RNA Viral Infection. Viruses 2022, 14, 613. https://doi.org/10.3390/v14030613
Feng M, Zhang H. Aminoacyl-tRNA Synthetase: A Non-Negligible Molecule in RNA Viral Infection. Viruses. 2022; 14(3):613. https://doi.org/10.3390/v14030613
Chicago/Turabian StyleFeng, Min, and Han Zhang. 2022. "Aminoacyl-tRNA Synthetase: A Non-Negligible Molecule in RNA Viral Infection" Viruses 14, no. 3: 613. https://doi.org/10.3390/v14030613
APA StyleFeng, M., & Zhang, H. (2022). Aminoacyl-tRNA Synthetase: A Non-Negligible Molecule in RNA Viral Infection. Viruses, 14(3), 613. https://doi.org/10.3390/v14030613