
On the Origin and Propagation of the COVID-19 Outbreak in the Italian Province of Trento, a Tourist Region of Northern Italy

 ,
,  ,
,  , , , , , , ,
, , , , , , ,  ,
,  , and
, and 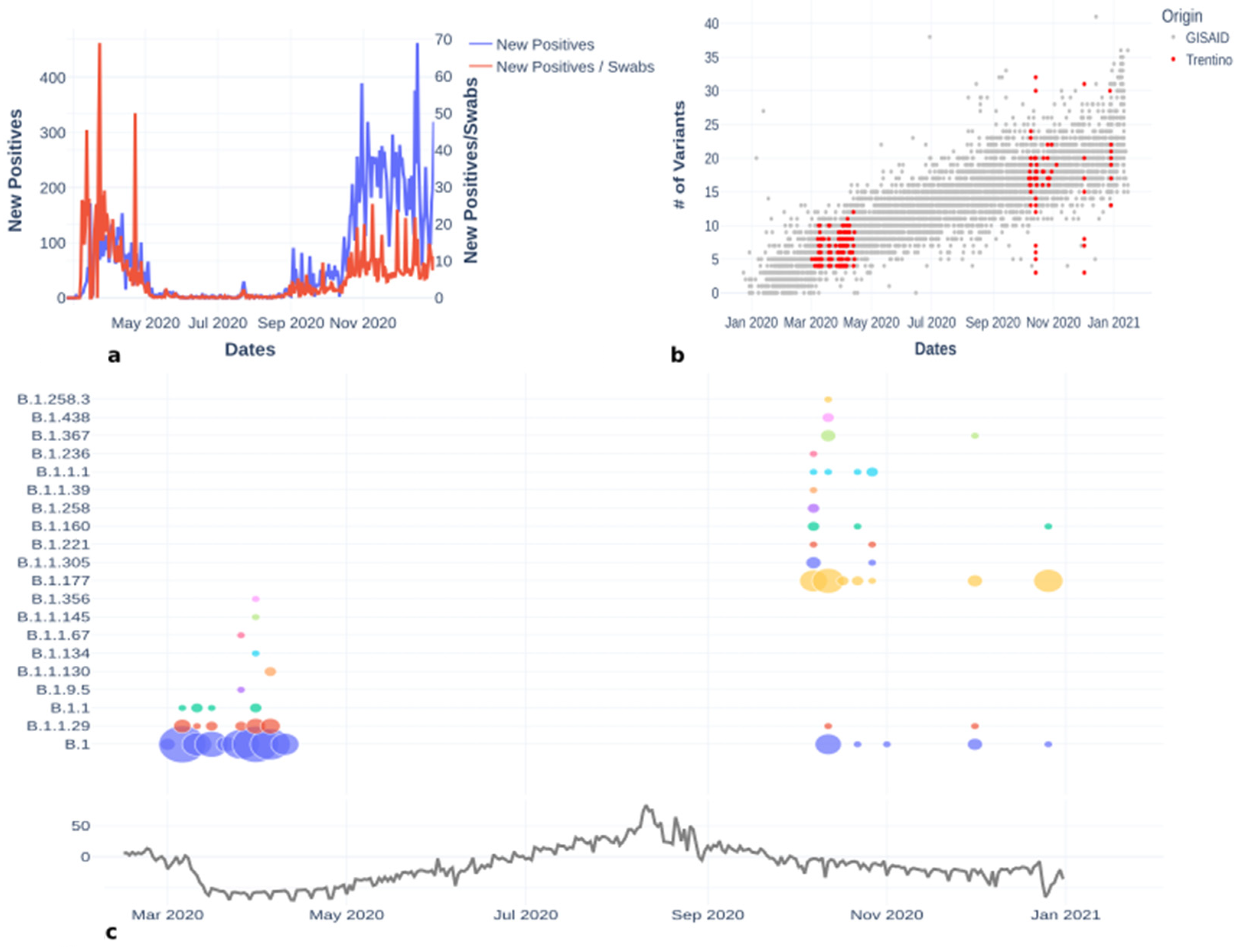{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Samples
2.2. Sequencing
2.3. Quality of the Sequenced Genomes
2.4. Phylogenetic Analysis
2.4.1. Data Preparation and Retrieval
2.4.2. Sequence Alignment, Data Cleaning and Homoplasy Analysis
2.4.3. Bayesian Molecular Clock Analysis
2.5. Minimum Spanning Networks
2.5.1. Selection of Sequences from GISAID
2.5.2. Construction of the Network
2.6. Mobility Data Processing
3. Results
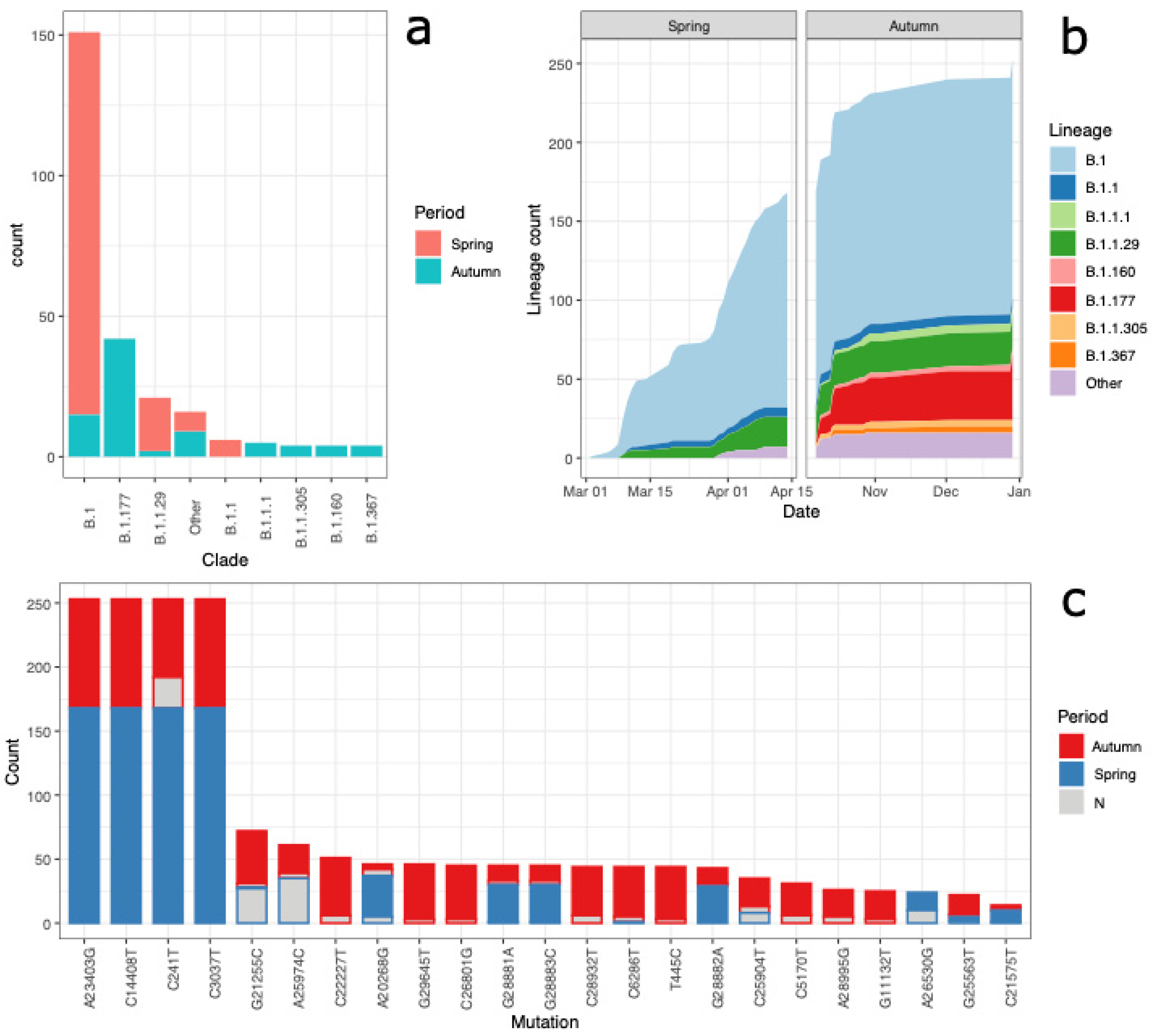
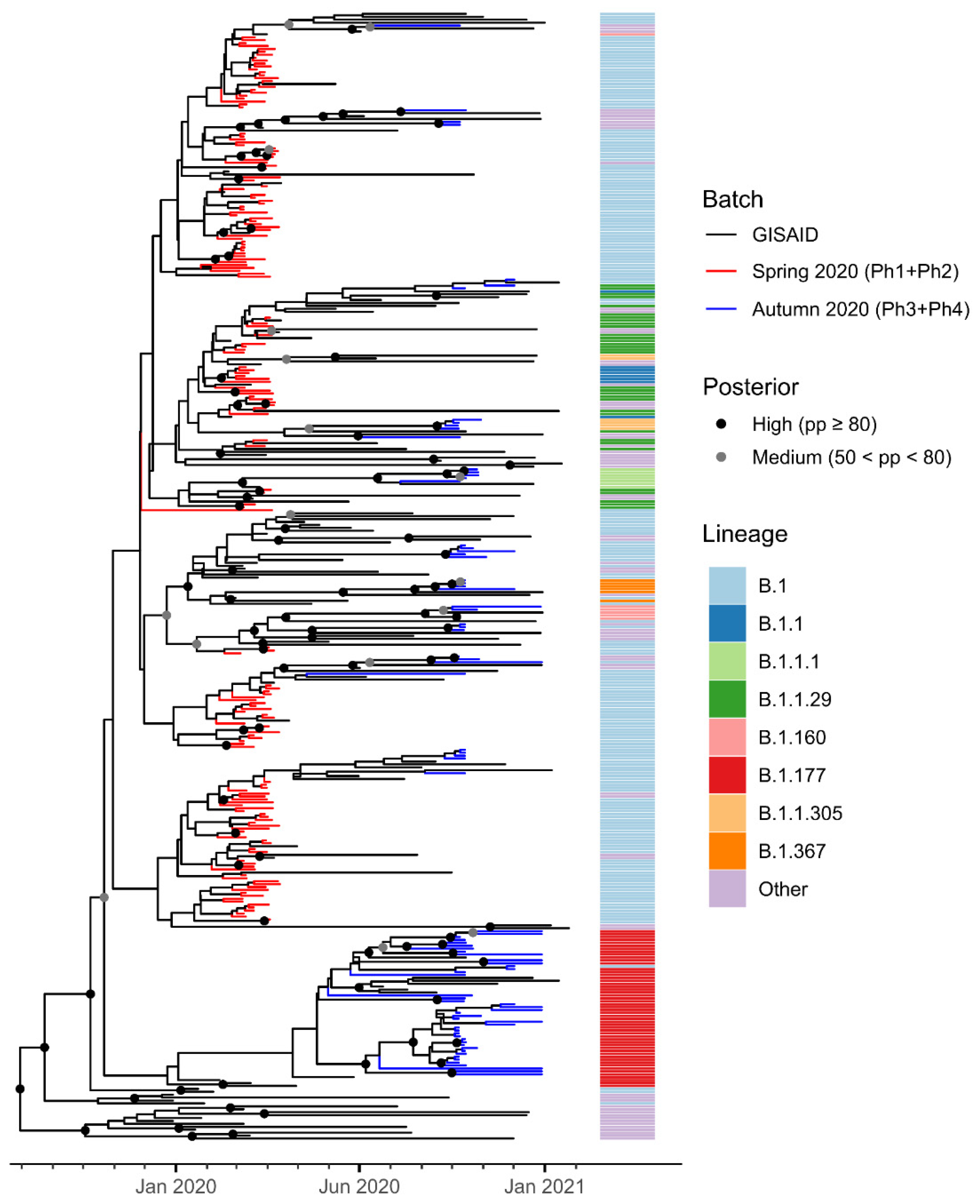
3.1. Lineages of the First and Second Wave
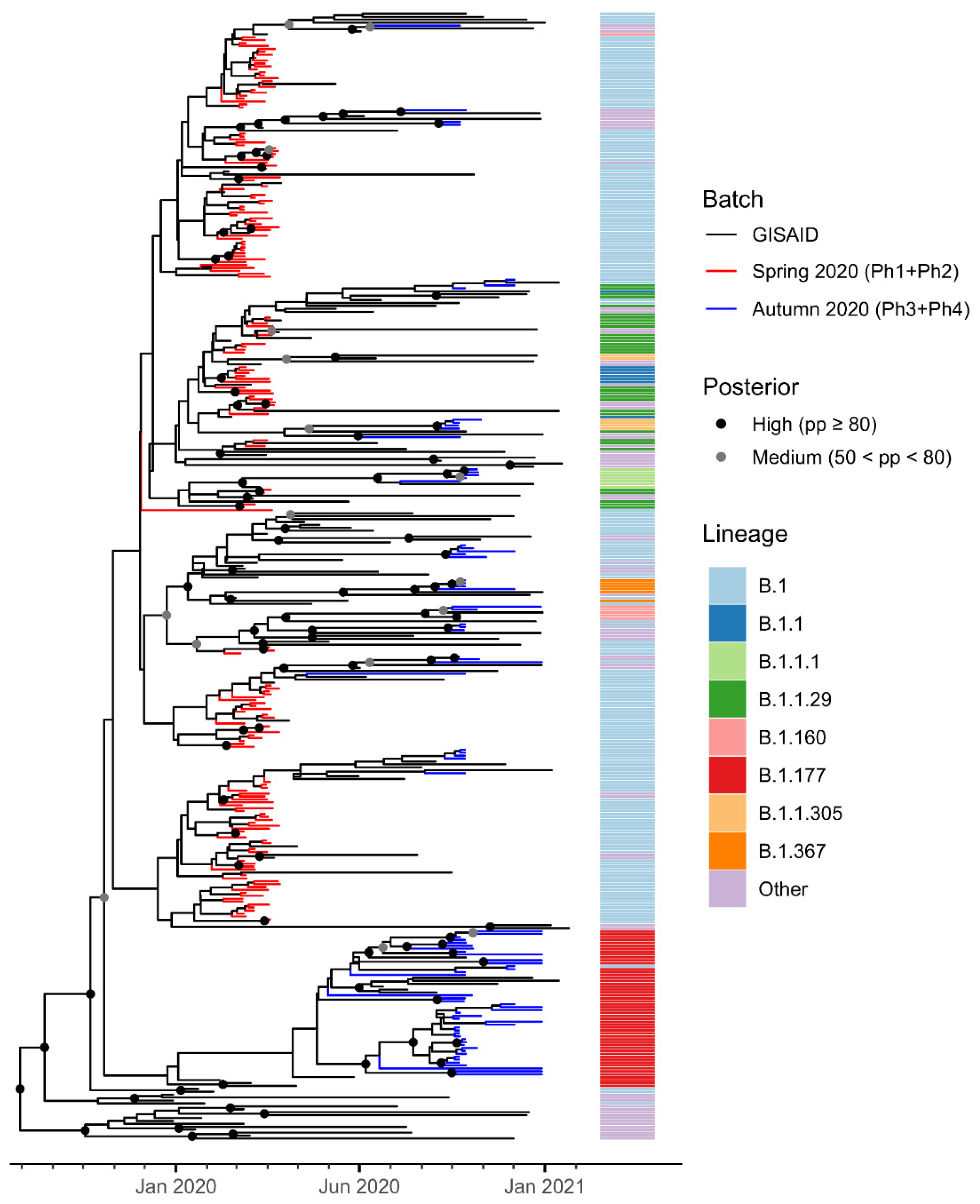
3.2. Phylogenetic Analysis
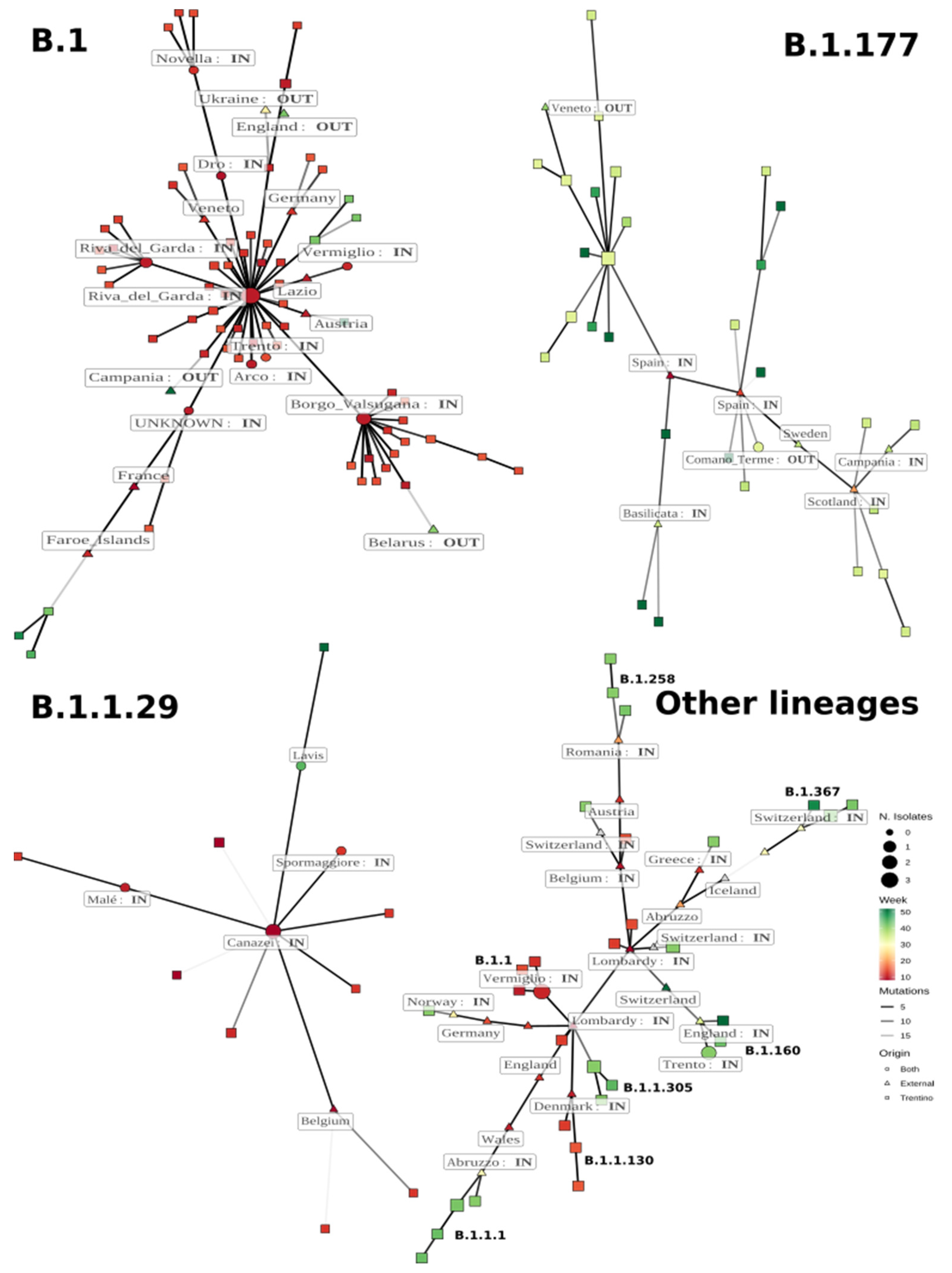
3.3. Minimum Spanning Network Analysis
3.3.1. Lineage B.1
3.3.2. Lineage B.1.177
3.3.3. Lineage B.1.1.29
3.3.4. Other Lineages Detected in Trentino
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinceti, M.; Filippini, T.; Rothman, K.J.; Di Federico, S.; Orsini, N. SARS-CoV-2 Infection Incidence during the First and Second COVID-19 Waves in Italy. Environ. Res. 2021, 197, 111097. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Vinh, L.S.; Flouri, T.; Stamatakis, A.; von Haeseler, A.; Minh, B.Q. MPBoot: Fast Phylogenetic Maximum Parsimony Tree Inference and Bootstrap Approximation. BMC Evol. Biol. 2018, 18, 11. [Google Scholar] [CrossRef] [Green Version]
- Crispell, J.; Balaz, D.; Gordon, S.V. HomoplasyFinder: A Simple Tool to Identify Homoplasies on a Phylogeny. Microb. Genom. 2019, 5, e000245. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alteri, C.; Cento, V.; Piralla, A.; Costabile, V.; Tallarita, M.; Colagrossi, L.; Renica, S.; Giardina, F.; Novazzi, F.; Gaiarsa, S.; et al. Genomic Epidemiology of SARS-CoV-2 Reveals Multiple Lineages and Early Spread of SARS-CoV-2 Infections in Lombardy, Italy. Nat. Commun. 2021, 12, 434. [Google Scholar] [CrossRef] [PubMed]
- Pekar, J.; Worobey, M.; Moshiri, N.; Scheffler, K.; Wertheim, J.O. Timing the SARS-CoV-2 Index Case in Hubei Province. Science 2021, 372, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Duchene, S.; Lemey, P.; Stadler, T.; Ho, S.Y.W.; Duchene, D.A.; Dhanasekaran, V.; Baele, G. Bayesian Evaluation of Temporal Signal in Measurably Evolving Populations. Mol. Biol. Evol. 2020, 37, 3363–3379. [Google Scholar] [CrossRef] [PubMed]
- Pipes, L.; Wang, H.; Huelsenbeck, J.P.; Nielsen, R. Assessing Uncertainty in the Rooting of the SARS-CoV-2 Phylogeny. Mol. Biol. Evol. 2021, 38, 1537–1543. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. GGTREE: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Vinceti, M.; Filippini, T.; Rothman, K.J.; Ferrari, F.; Goffi, A.; Maffeis, G.; Orsini, N. Lockdown Timing and Efficacy in Controlling COVID-19 Using Mobile Phone Tracking. EClinicalMedicine 2020, 25, 100457. [Google Scholar] [CrossRef]
- Fountain-Jones, N.M.; Appaw, R.C.; Carver, S.; Didelot, X.; Volz, E.; Charleston, M. Emerging Phylogenetic Structure of the SARS-CoV-2 Pandemic. Virus Evol. 2020, 6, veaa082. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 Variant through Europe in the Summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Di Giallonardo, F.; Duchene, S.; Puglia, I.; Curini, V.; Profeta, F.; Cammà, C.; Marcacci, M.; Calistri, P.; Holmes, E.; Lorusso, A. Genomic Epidemiology of the First Wave of SARS-CoV-2 in Italy. Viruses 2020, 12, 1438. [Google Scholar] [CrossRef] [PubMed]
- Engelbrecht, S.; Delaney, K.; Kleinhans, B.; Wilkinson, E.; Tegally, H.; Stander, T.; van Zyl, G.; Preiser, W.; de Oliveira, T. Multiple Early Introductions of SARS-CoV-2 to Cape Town, South Africa. Viruses 2021, 13, 526. [Google Scholar] [CrossRef] [PubMed]
- Yi, B.; Poetsch, A.R.; Stadtmüller, M.; Rost, F.; Winkler, S.; Dalpke, A.H. Phylogenetic analysis of SARS-CoV-2 lineage development across the first and second waves in Eastern Germany in 2020: Insights into the cause of the second wave. Epidemiol. Infect. 2021, 149, E177. [Google Scholar] [CrossRef]
- Chrysostomou, A.C.; Vrancken, B.; Koumbaris, G.; Themistokleous, G.; Aristokleous, A.; Masia, C.; Eleftheriou, C.; Iοannou, C.; Stylianou, D.C.; Ioannides, M.; et al. A Comprehensive Molecular Epidemiological Analysis of SARS-CoV-2 Infection in Cyprus from April 2020 to January 2021: Evidence of a Highly Polyphyletic and Evolving Epidemic. Viruses 2021, 13, 1098. [Google Scholar] [CrossRef]
- La Rosa, G.; Mancini, P.; Bonanno Ferraro, G.; Veneri, C.; Iaconelli, M.; Bonadonna, L.; Lucentini, L.; Suffredini, E. SARS-CoV-2 Has Been Circulating in Northern Italy since December 2019: Evidence from Environmental Monitoring. Sci. Total Environ. 2021, 750, 141711. [Google Scholar] [CrossRef]
- Amendola, A.; Bianchi, S.; Gori, M.; Colzani, D.; Canuti, M.; Borghi, E.; Raviglione, M.C.; Zuccotti, G.V.; Tanzi, E. Evidence of SARS-CoV-2 RNA in an Oropharyngeal Swab Specimen, Milan, Italy, Early December 2019. Emerg. Infect. Dis. 2021, 27, 648–650. [Google Scholar] [CrossRef]
- Lemey, P.; Ruktanonchai, N.; Hong, S.L.; Colizza, V.; Poletto, C.; Van den Broeck, F.; Gill, M.S.; Ji, X.; Levasseur, A.; Oude Munnink, B.B.; et al. Untangling Introductions and Persistence in COVID-19 Resurgence in Europe. Nature 2021, 595, 713–717. [Google Scholar] [CrossRef]
- Lavezzo, E.; Franchin, E.; Ciavarella, C.; Cuomo-Dannenburg, G.; Barzon, L.; Del Vecchio, C.; Rossi, L.; Manganelli, R.; Loregian, A.; Navarin, N.; et al. Suppression of a SARS-CoV-2 Outbreak in the Italian Municipality of Vo’. Nature 2020, 584, 425–429. [Google Scholar] [CrossRef]
- Morel, B.; Barbera, P.; Czech, L.; Bettisworth, B.; Hübner, L.; Lutteropp, S.; Serdari, D.; Kostaki, E.-G.; Mamais, I.; Kozlov, A.M.; et al. Phylogenetic Analysis of SARS-CoV-2 Data Is Difficult. Mol. Biol. Evol. 2021, 38, 1777–1791. [Google Scholar] [CrossRef]
- Lu, J.; du Plessis, L.; Liu, Z.; Hill, V.; Kang, M.; Lin, H.; Sun, J.; François, S.; Kraemer, M.U.G.; Faria, N.R.; et al. Genomic Epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell 2020, 181, 997.e9–1003.e9. [Google Scholar] [CrossRef] [PubMed]
- Manuto, L.; Grazioli, M.; Spitaleri, A.; Fontana, P.; Bianco, L.; Bertolotti, L.; Bado, M.; Mazzotti, G.; Bianca, F.; Onelia, F.; et al. Rapid SARS-CoV-2 Intra-Host and Within-Household Emergence of Novel Haplotypes. Viruses 2022, 14, 399. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianco, L.; Moser, M.; Silverj, A.; Micheletti, D.; Lorenzin, G.; Collini, L.; Barbareschi, M.; Lanzafame, P.; Segata, N.; Pindo, M.; et al. On the Origin and Propagation of the COVID-19 Outbreak in the Italian Province of Trento, a Tourist Region of Northern Italy. Viruses 2022, 14, 580. https://doi.org/10.3390/v14030580
Bianco L, Moser M, Silverj A, Micheletti D, Lorenzin G, Collini L, Barbareschi M, Lanzafame P, Segata N, Pindo M, et al. On the Origin and Propagation of the COVID-19 Outbreak in the Italian Province of Trento, a Tourist Region of Northern Italy. Viruses. 2022; 14(3):580. https://doi.org/10.3390/v14030580
Chicago/Turabian StyleBianco, Luca, Mirko Moser, Andrea Silverj, Diego Micheletti, Giovanni Lorenzin, Lucia Collini, Mattia Barbareschi, Paolo Lanzafame, Nicola Segata, Massimo Pindo, and et al. 2022. "On the Origin and Propagation of the COVID-19 Outbreak in the Italian Province of Trento, a Tourist Region of Northern Italy" Viruses 14, no. 3: 580. https://doi.org/10.3390/v14030580
APA StyleBianco, L., Moser, M., Silverj, A., Micheletti, D., Lorenzin, G., Collini, L., Barbareschi, M., Lanzafame, P., Segata, N., Pindo, M., Franceschi, P., Rota-Stabelli, O., Rizzoli, A., Fontana, P., & Donati, C. (2022). On the Origin and Propagation of the COVID-19 Outbreak in the Italian Province of Trento, a Tourist Region of Northern Italy. Viruses, 14(3), 580. https://doi.org/10.3390/v14030580