
Restriction-Assembly: A Solution to Construct Novel Adenovirus Vector

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus, Plasmids and Oligonucleotides
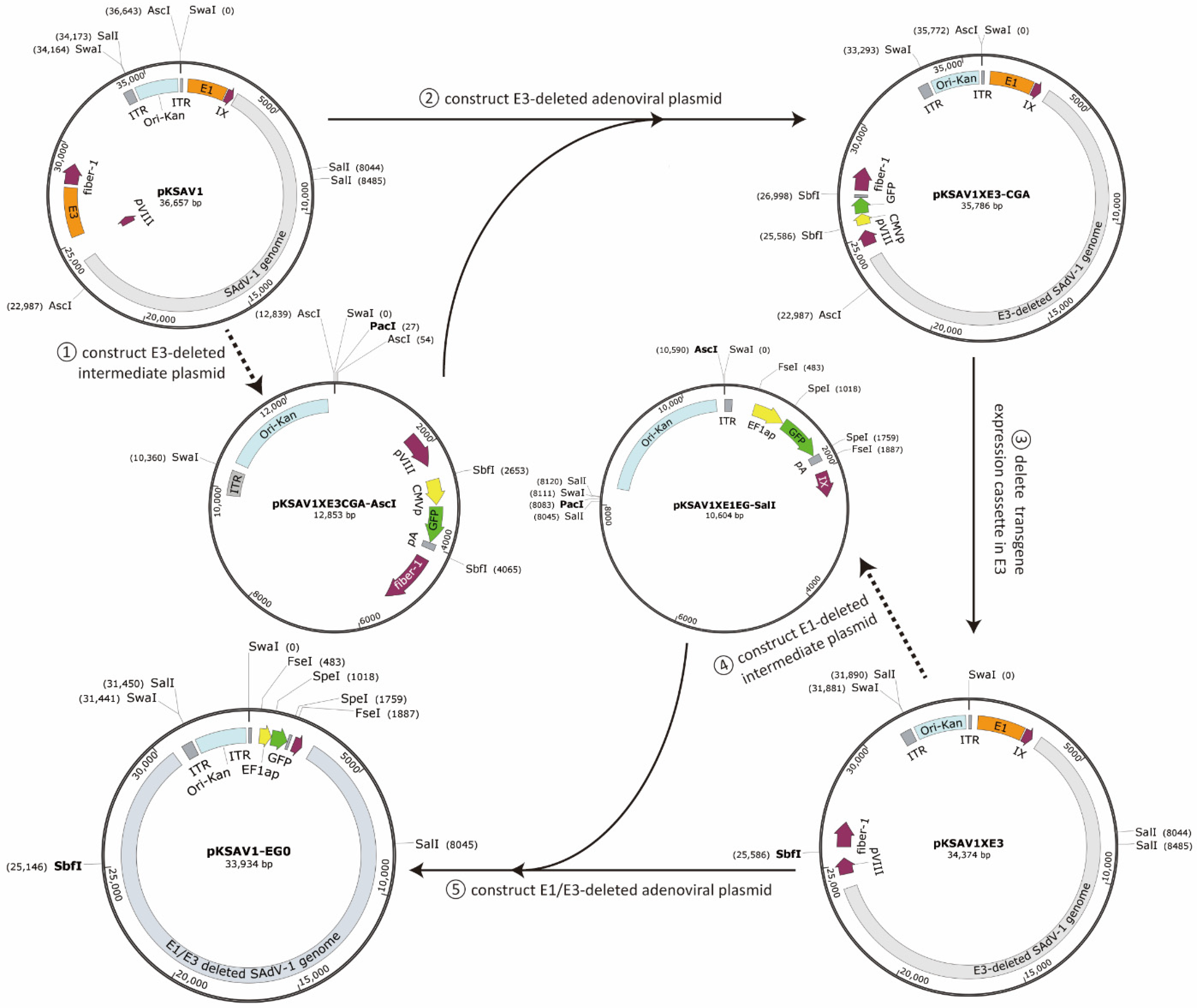
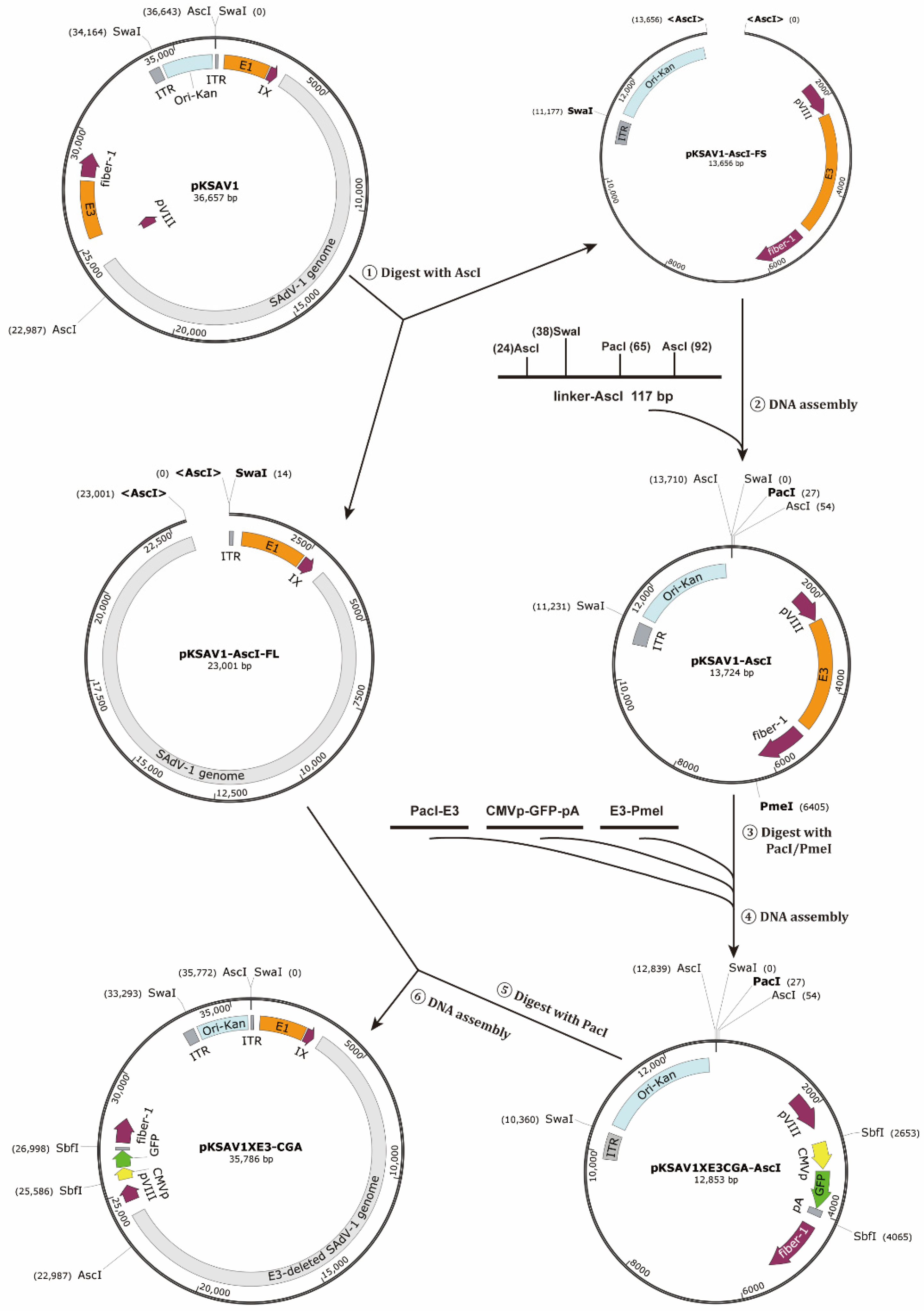
2.2. Construction of E3-Deleted SAdV-1 Vector
2.3. Construction of E1/E3-Deleted SAdV-1 Vector
2.4. Rescue, Amplification, Purification and Titration of Recombinant Virus
2.5. Establishment of 293 Cell Lines Stably Expressing SAdV-1 E1B55K Gene
2.6. Transduction of Cell Lines
2.7. Adenovirus Neutralization Assay
3. Results
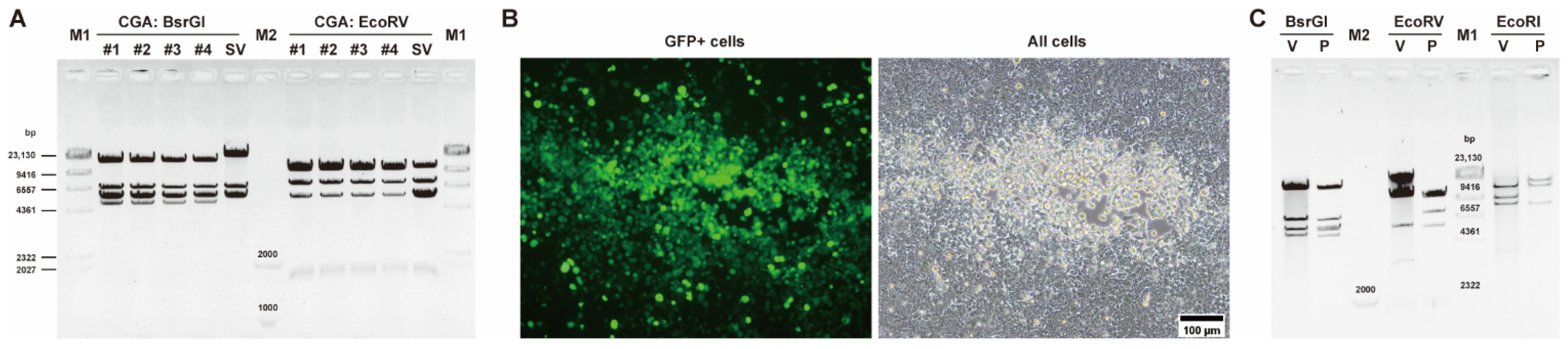
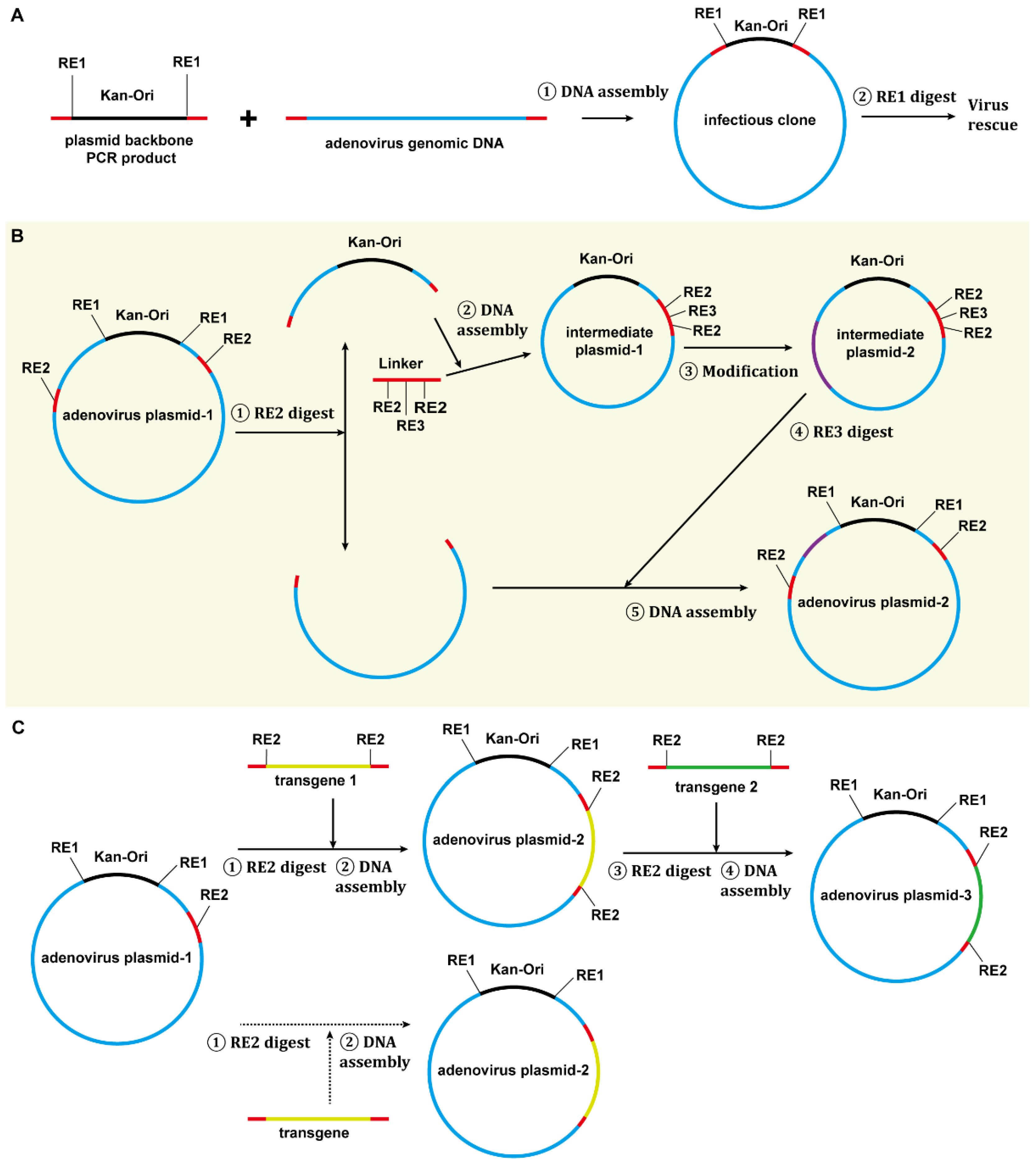
3.1. Construction of E3-Deleted SAdV-1 Vector
3.2. Construction of E1/E3-Deleted SAdV-1 Adenoviral Plasmid
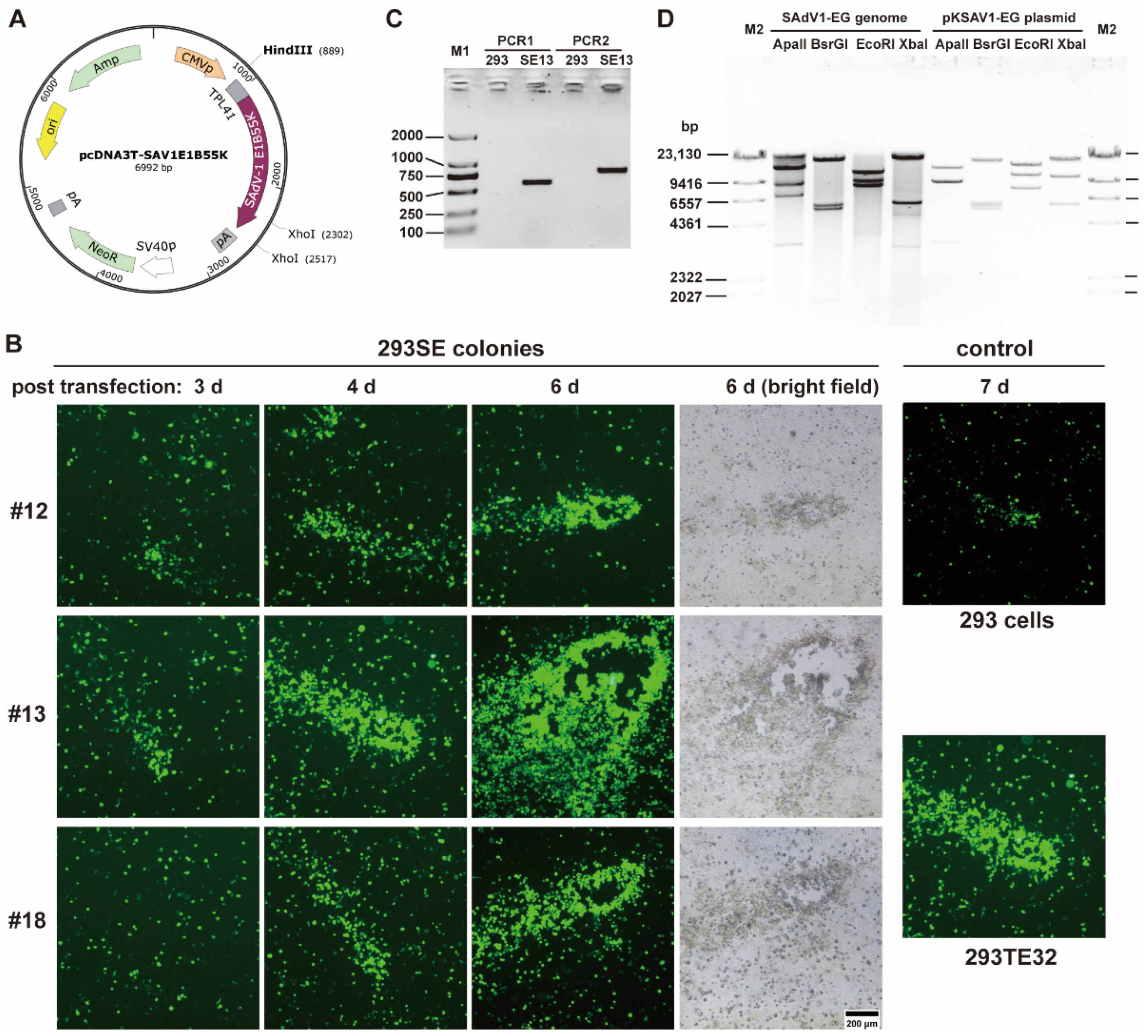
3.3. Establishment of SAdV-1 E1B55K-Integrated 293 Cells
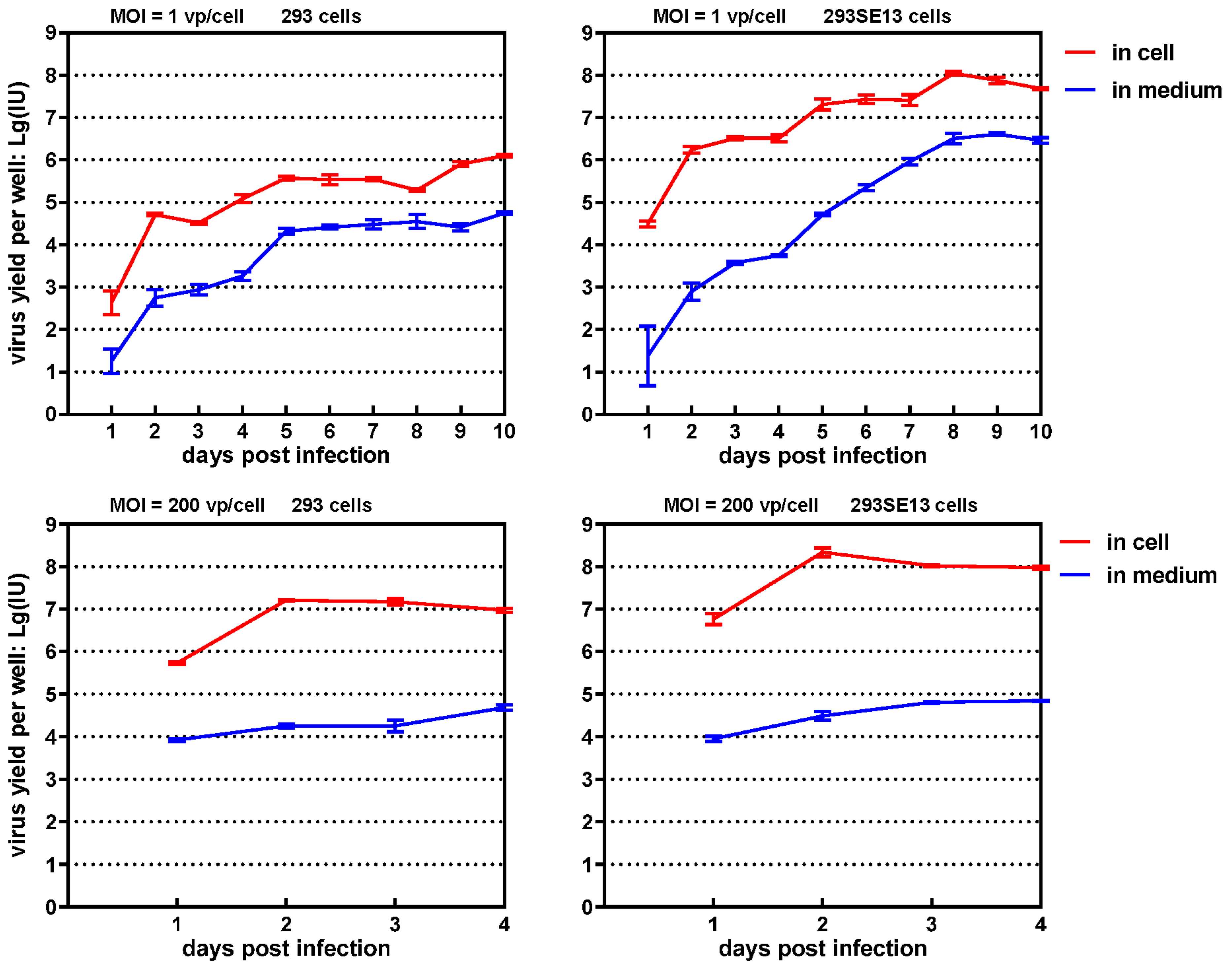
3.4. One-Step Growth Curve of SAdV1-EG
3.5. Gene Transduction Efficiency of SAdV1-EG
3.6. Replication of E3-Deleted SAdV-1 in K562 Cells
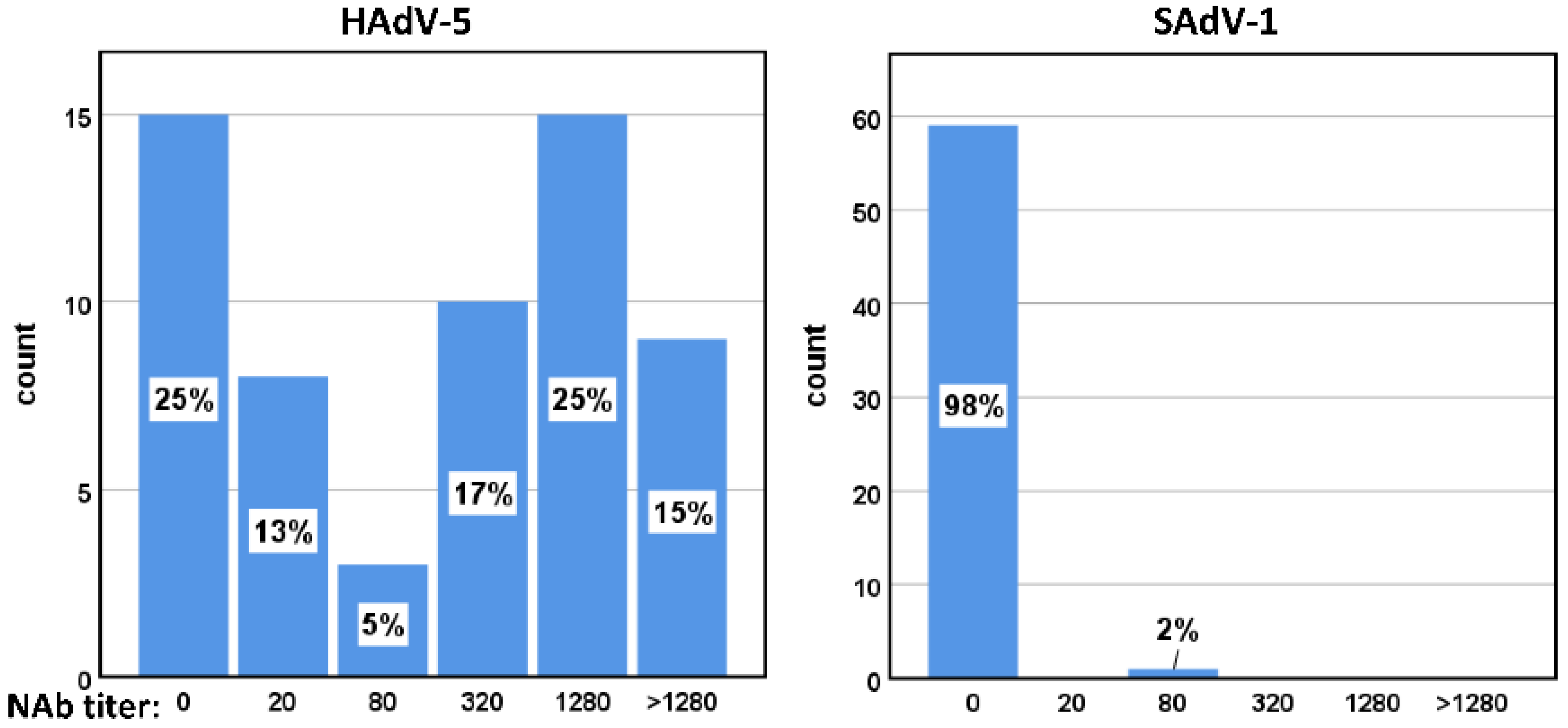
3.7. Seroprevalence of HAdV-5 and SAdV-1 Neutralizing Antibodies in Healthy Adults
4. Discussion
4.1. There Is a Need to Construct Novel Adenovirus Vectors
4.2. The Application of Restriction-Assembly in Constructing Adenovirus Vectors
4.3. Advantages of Restriction-Assembly
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Virus Taxonomy; Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier Academic Press: San Diego, CA, USA, 2011; pp. 125–141. [Google Scholar]
- Ji, T.; Li, L.; Li, W.; Zheng, X.; Ye, X.; Chen, H.; Zhou, Q.; Jia, H.; Chen, B.; Lin, Z.; et al. Emergence and characterization of a putative novel human adenovirus recombinant HAdV-C104 causing pneumonia in Southern China. Virus Evol. 2021, 7, veab018. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G. Adenovirus: The first effective in vivo gene delivery vector. Hum. Gene Ther. 2014, 25, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Nagasato, M.; Yoshida, T.; Aoki, K. Recent advances in genetic modification of adenovirus vectors for cancer treatment. Cancer Sci. 2017, 108, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, S.P.; Mehrotra, D.V.; Duerr, A.; Fitzgerald, D.W.; Mogg, R.; Li, D.; Gilbert, P.B.; Lama, J.R.; Marmor, M.; Del Rio, C.; et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 2008, 372, 1881–1893. [Google Scholar] [CrossRef] [Green Version]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against Ad vectors: Humoral, cellular, and innate response, what’s important? Hum. Vaccin. Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.X.; Zou, X.H.; Jiang, S.Y.; Lu, N.N.; Han, M.; Zhao, J.H.; Guo, X.J.; Zhao, S.C.; Lu, Z.Z. Prevalence of serum neutralizing antibodies to adenovirus type 5 (Ad5) and 41 (Ad41) in children is associated with age and sanitary conditions. Vaccine 2016, 34, 5579–5586. [Google Scholar] [CrossRef]
- Mennechet, F.J.D.; Paris, O.; Ouoba, A.R.; Salazar Arenas, S.; Sirima, S.B.; Takoudjou Dzomo, G.R.; Diarra, A.; Traore, I.T.; Kania, D.; Eichholz, K.; et al. A review of 65 years of human adenovirus seroprevalence. Expert Rev. Vaccines 2019, 18, 597–613. [Google Scholar] [CrossRef]
- Lopez-Gordo, E.; Podgorski, I.I.; Downes, N.; Alemany, R. Circumventing antivector immunity: Potential use of nonhuman adenoviral vectors. Hum. Gene Ther. 2014, 25, 285–300. [Google Scholar] [CrossRef] [Green Version]
- Vitelli, A.; Folgori, A.; Scarselli, E.; Colloca, S.; Capone, S.; Nicosia, A. Chimpanzee adenoviral vectors as vaccines—challenges to move the technology into the fast lane. Expert Rev. Vaccines 2017, 16, 1241–1252. [Google Scholar] [CrossRef]
- Abbink, P.; Kirilova, M.; Boyd, M.; Mercado, N.; Li, Z.; Nityanandam, R.; Nanayakkara, O.; Peterson, R.; Larocca, R.A.; Aid, M.; et al. Rapid Cloning of Novel Rhesus Adenoviral Vaccine Vectors. J. Virol. 2018, 92, e01924-17. [Google Scholar] [CrossRef] [Green Version]
- Sasso, E.; D’Alise, A.M.; Zambrano, N.; Scarselli, E.; Folgori, A.; Nicosia, A. New viral vectors for infectious diseases and cancer. Semin. Immunol. 2020, 50, 101430. [Google Scholar] [CrossRef] [PubMed]
- Chang, J. Adenovirus Vectors: Excellent Tools for Vaccine Development. Immune Netw. 2021, 21, e6. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Mese, K.; Bunz, O.; Ehrhardt, A. State-of-the-art human adenovirus vectorology for therapeutic approaches. FEBS Lett. 2019, 593, 3609–3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanerva, A.; Hemminki, A. Modified adenoviruses for cancer gene therapy. Int. J. Cancer 2004, 110, 475–480. [Google Scholar] [CrossRef]
- Muck-Hausl, M.; Solanki, M.; Zhang, W.; Ruzsics, Z.; Ehrhardt, A. Ad 2.0: A novel recombineering platform for high-throughput generation of tailored adenoviruses. Nucleic Acids Res. 2015, 43, e50. [Google Scholar] [CrossRef]
- Chartier, C.; Degryse, E.; Gantzer, M.; Dieterle, A.; Pavirani, A.; Mehtali, M. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J. Virol. 1996, 70, 4805–4810. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Mondal, M.; Zhou, D. Development of novel vaccine vectors: Chimpanzee adenoviral vectors. Hum. Vaccin. Immunother. 2018, 14, 1679–1685. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chi, Y.; Tang, X.; Ertl, H.C.J.; Zhou, D. Rapid, Efficient, and Modular Generation of Adenoviral Vectors via Isothermal Assembly. Curr. Protoc. Mol. Biol. 2016, 113, 16–26. [Google Scholar] [CrossRef]
- Luo, S.; Zhang, P.; Ma, X.; Wang, Q.; Lu, J.; Liu, B.; Zhao, W.; Allain, J.P.; Li, C.; Li, T. A rapid strategy for constructing novel simian adenovirus vectors with high viral titer and expressing highly antigenic proteins applicable for vaccine development. Virus Res. 2019, 268, 1–10. [Google Scholar] [CrossRef]
- Guo, X.; Mei, L.; Yan, B.; Zou, X.; Hung, T.; Lu, Z. Site-directed modification of adenoviral vector with combined DNA assembly and restriction-ligation cloning. J. Biotechnol. 2020, 307, 193–201. [Google Scholar] [CrossRef]
- Zou, X.H.; Xiao, X.; Chen, D.L.; Li, Z.L.; Song, J.D.; Wang, M.; Qu, J.G.; Lu, Z.Z.; Hung, T. An improved HAdV-41 E1B55K-expressing 293 cell line for packaging fastidious adenovirus. J. Virol. Methods 2011, 175, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.M.; Harrach, B.; Zakhartchouk, A.N.; Davison, A.J. Complete genome sequence of simian adenovirus 1: An Old World monkey adenovirus with two fiber genes. J. Gen. Virol. 2005, 86, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Arad, U. Modified Hirt procedure for rapid purification of extrachromosomal DNA from mammalian cells. BioTechniques 1998, 24, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.Z.; Zou, X.H.; Dong, L.X.; Qu, J.G.; Song, J.D.; Wang, M.; Guo, L.; Hung, T. Novel recombinant adenovirus type 41 vector and its biological properties. J. Gene Med. 2009, 11, 128–138. [Google Scholar] [CrossRef]
- Lv, Y.; Xiao, F.J.; Wang, Y.; Zou, X.H.; Wang, H.; Wang, H.Y.; Wang, L.S.; Lu, Z.Z. Efficient gene transfer into T lymphocytes by fiber-modified human adenovirus 5. BMC Biotechnol. 2019, 19, 23. [Google Scholar] [CrossRef]
- Yan, B.; Zou, X.; Liu, X.; Zhao, J.; Zhang, W.; Guo, X.; Wang, M.; Lv, Y.; Lu, Z. User-Friendly Reverse Genetics System for Modification of the Right End of Fowl Adenovirus 4 Genome. Viruses 2020, 12, 301. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Rong, Y.; Guo, X.; Hou, W.; Yan, B.; Hung, T.; Lu, Z. Fiber1, but not fiber2, is the essential fiber gene for fowl adenovirus 4 (FAdV-4). J. Gen. Virol. 2021, 102. [Google Scholar] [CrossRef]
- Zou, X.H.; Bi, Z.X.; Guo, X.J.; Zhang, Z.; Zhao, Y.; Wang, M.; Zhu, Y.L.; Jie, H.Y.; Yu, Y.; Hung, T.; et al. DNA assembly technique simplifies the construction of infectious clone of fowl adenovirus. J. Virol. Methods 2018, 257, 85–92. [Google Scholar] [CrossRef]
- Davis, A.R.; Wivel, N.A.; Palladino, J.L.; Tao, L.; Wilson, J.M. Construction of adenoviral vectors. Methods Mol. Biol. 2000, 135, 515–523. [Google Scholar] [CrossRef]
- Chen, D.L.; Dong, L.X.; Li, M.; Guo, X.J.; Wang, M.; Liu, X.F.; Lu, Z.Z.; Hung, T. Construction of an infectious clone of human adenovirus type 41. Arch. Virol. 2012, 157, 1313–1321. [Google Scholar] [CrossRef]
- McFarland, D.C. Preparation of pure cell cultures by cloning. Methods Cell Sci. 2000, 22, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Vogels, R.; Zuijdgeest, D.; van Rijnsoever, R.; Hartkoorn, E.; Damen, I.; de Bethune, M.P.; Kostense, S.; Penders, G.; Helmus, N.; Koudstaal, W.; et al. Replication-deficient human adenovirus type 35 vectors for gene transfer and vaccination: Efficient human cell infection and bypass of preexisting adenovirus immunity. J. Virol. 2003, 77, 8263–8271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holterman, L.; Vogels, R.; van der Vlugt, R.; Sieuwerts, M.; Grimbergen, J.; Kaspers, J.; Geelen, E.; van der Helm, E.; Lemckert, A.; Gillissen, G.; et al. Novel replication-incompetent vector derived from adenovirus type 11 (Ad11) for vaccination and gene therapy: Low seroprevalence and non-cross-reactivity with Ad5. J. Virol. 2004, 78, 13207–13215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.Z.; Ni, F.; Hu, Z.B.; Wang, L.; Wang, H.; Zhang, Q.W.; Huang, W.R.; Wu, C.T.; Wang, L.S. Efficient gene transfer into hematopoietic cells by a retargeting adenoviral vector system with a chimeric fiber of adenovirus serotype 5 and 11p. Exp. Hematol. 2006, 34, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Arnberg, N. Adenovirus receptors: Implications for targeting of viral vectors. Trends Pharmacol. Sci. 2012, 33, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.L.; Cannon, P.M. Promoter choice for retroviral vectors: Transcriptional strength versus trans-activation potential. Hum. Gene Ther. 2007, 18, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Gomez-Gonzalez, A. Adenovirus—A blueprint for gene delivery. Curr. Opin Virol. 2021, 48, 49–56. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, W.; Ehrhardt, A. Expanding the Spectrum of Adenoviral Vectors for Cancer Therapy. Cancers 2020, 12, 1139. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Z.; Li, C.; Gil, S.; Papayannopoulou, T.; Doering, C.B.; Lieber, A. High-level protein production in erythroid cells derived from in vivo transduced hematopoietic stem cells. Blood Adv. 2019, 3, 2883–2894. [Google Scholar] [CrossRef]
- Hausl, M.; Zhang, W.; Voigtlander, R.; Muther, N.; Rauschhuber, C.; Ehrhardt, A. Development of adenovirus hybrid vectors for Sleeping Beauty transposition in large mammals. Curr. Gene Ther. 2011, 11, 363–374. [Google Scholar] [CrossRef]
- Coughlan, L. Factors Which Contribute to the Immunogenicity of Non-replicating Adenoviral Vectored Vaccines. Front. Immunol. 2020, 11, 909. [Google Scholar] [CrossRef] [PubMed]
- Arnberg, N. Adenovirus receptors: Implications for tropism, treatment and targeting. Rev. Med. Virol. 2009, 19, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Francois, A.; Eterradossi, N.; Delmas, B.; Payet, V.; Langlois, P. Construction of avian adenovirus CELO recombinants in cosmids. J. Virol. 2001, 75, 5288–5301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, M.; Ljungberg, J.; Richter, J.; Kiefer, T.; Magnusson, M.; Lieber, A.; Widegren, B.; Karlsson, S.; Fan, X. Development of an adenoviral vector system with adenovirus serotype 35 tropism; efficient transient gene transfer into primary malignant hematopoietic cells. J. Gene Med. 2004, 6, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Xiong, A.S.; Yao, Q.H.; Peng, R.H.; Li, X.; Fan, H.Q.; Cheng, Z.M.; Li, Y. A simple, rapid, high-fidelity and cost-effective PCR-based two-step DNA synthesis method for long gene sequences. Nucleic Acids Res. 2004, 32, e98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Lu, Z.; Zhang, X.; Guo, X.; Mei, L.; Zou, X.; Zhong, Y.; Wang, M.; Hung, T. Single Plasmid-Based, Upgradable, and Backward-Compatible Adenoviral Vector Systems. Hum. Gene Ther. 2019, 30, 777–791. [Google Scholar] [CrossRef] [Green Version]
- He, T.C.; Zhou, S.; da Costa, L.T.; Yu, J.; Kinzler, K.W.; Vogelstein, B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 1998, 95, 2509–2514. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Collins, L.T.; Curiel, D.T. Synthetic Biology Approaches for Engineering Next-Generation Adenoviral Gene Therapies. ACS Nano 2021, 15, 13970–13979. [Google Scholar] [CrossRef]
- Kinoshita, T.; Imamura, J.; Nagai, H.; Shimotohno, K. Quantification of gene expression over a wide range by the polymerase chain reaction. Anal. Biochem. 1992, 206, 231–235. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragment | Oligo Name | Sequence | Template | Product (bp) | Restriction Enzyme |
|---|---|---|---|---|---|
| Kan-Ori | 2001KSAV1p1 | gtttccagaa taaggtatat tattgatgat gatttaaatc caagtcgac | pKFAV4GFP | 2514 | SwaI, SalI |
| 2001KSAV1p2 | tgatgatgat ttaaatccaa gtcgacgatc ccgagcggta tcagctc | ||||
| 2001KSAV1p3 | tgatgattta aatggttggc gcgcctggaa caacactcaa ccctatcg | 2563 | |||
| 2001KSAV1p4 | gtttccagaa taaggtatat tattgatgat gatttaaatg gttggcgcgc c | SwaI, AscI | |||
| AscI-linker | 2001KSAV1p5 | gatagggttg agtgttgttc caggcgcgcc aaccatttaa atcatcat | 117 | AscI, SwaI | |
| 2001KSAV1p6 | gcgcgccaac catttaaatc atcatcaata atatacctta attaagac | self-anneal | SwaI, PacI | ||
| 2001KSAV1p7 | cgccgctggc ggcagaggag tttgtcttaa ttaaggtata ttattgat | PacI | |||
| 2001KSAV1p8 | cgctgaaacc ggaccacagg gcgcgccgct ggcggcagag gagtttgt | AscI | |||
| PacI-E3 | 2001KSAV1p9 | cgcgccaacc atttaaatca tcatcaataa tatacctt | pKSAV1-ASCI | 2680 | |
| 2001KSAV1p10 | gttatgtaac gcctgcagga tgtaatccgg gcgtggggca g | SbfI | |||
| CMVp-GFP-pA | 2001KSAV1p11 | ggattacatc ctgcaggcgt tacataactt acggtaaatg | pKFAV4GFP | 568 | SbfI |
| 2001KSAV1p12 | accatggtgg ctagctctag cggatctgac ggttcactaa ac | ||||
| 2001KSAV1p13 | gatccgctag agctagccac catggtgagc aagggcg | 770 | |||
| 2001KSAV1p14 | caataaacaa gttagctagc ttagagtccg gacttgtaca gctcgtcc | ||||
| 2001KSAV1p15 | gtccggactc taagctagct aacttgttta ttgcagctta taatggttac | 163 | |||
| 2001KSAV1p16 | ccttaaaaat atccctgcag gtaagataca ttgatgagtt tggacaaacc ac | 1442 | SbfI | ||
| E3-PmeI | 2001KSAV1p17 | catcaatgta tcttacctgc agggatattt ttaaggtgta aatcaataat aaacttacc | pKSAV1-ASCI | 1522 | HindIII |
| 2001KSAV1p18 | cattttgcgt agtaatggga tctctgtagt ttaagcttaa cactccaagt gg | ||||
| SalI-SalI | 2001KSAV1p19 | gacgctccat ggcctcgtag aagtccacgg cgaagttgaa aaattg | pKSAV1 | 529 | PacI |
| 2001KSAV1p20 | aatcatcatc aataatatac cttattaatt aacgctttcc tagagaagtt ctcggatc | ||||
| SalI-linker | 2001KSAV1p21 | gcgttaatta ataaggtata ttattgatga tgatttaaat ccaagtcgac | self-anneal | 73 | |
| 2001KSAV1p22 | gtgagctgat accgctcggg atcgtcgact tggatttaaa tcatcatcaa | ||||
| AscI-E1A | 2001KSAV1p23 | cgcgataggg ttgagtgttg ttccagg | pKSAV1 | 530 | |
| 2001KSAV1p24 | gagcggccgg cccgcggcag cgcggaggag aaaac | FseI | |||
| EF1ap-GFP-pA | 2001KSAV1p25 | cgctgccgcg ggccggccgc tccggtgccc gtcagtggg | pCDH-CMV-MCS-EF1-copGFP | 566 | FseI |
| 2001KSAV1p26 | catggtggc actagtgtag gcgccggtca c | SpeI | |||
| 2003SAV1EGFPf | gtgaccggcg cctacactag tgccaccatg gtgagcaagg g | pKSAV1E3CGA | 794 | SpeI | |
| 2003SAV2EGFPr | ggtcaaggaa ggcacggggg agactagttt agagtccgga cttgtacagc tc | SpeI | |||
| 2001KSAV1p29 | gactctaaac tagtctcccc cgtgccttcc ttgacc | pcDNA3 | 151 | SpeI | |
| 2001KSAV1p30 | ggatacaacc tcggccggcc accccacccc ccagaataga atg | FseI | |||
| E1B-EcoRV | 2001KSAV1p31 | ggtggccggc cgaggttgta tcctgtaacc ctgaacgt | pKSAV1 | 579 | FseI |
| 2001KSAV1p32 | tctgaagcgg tatcggggtt agcttgggat | ||||
| AsiSI-RITR | 2010SAdV1RITR1 | taacagaccc aggtcaggtt gctctc | pKSAV1-EG0/SphI-SnaBI | 997 | |
| 2010SAdV1RITR2 | cgagggcggg cgggcgaagg gcgtgtgtgg gaaag | ||||
| RITR-AseI | 2010SAdV1RITR3 | ccttcgcccg cccgccctcg cgccaccccg cgtca | pKSAV1-EG0/SphI-SnaBI | 1324 | |
| 2010SAdV1RITR4 | cgaactactt actctagctt cccggcaaca | ||||
| SAdV1-E1B55K | 2007SAV1E1Bf | gctgccttta ttacctatat tttgg | SAdV-1 genome | 1489 | |
| 2007SAV1E1Br | cctcatgccc ctttataccc tt | ||||
| 2007TSAV1E1Bf | tcgagccaat cacagtcgca agatggagca acagcgacag cc | 1466 | |||
| 2007TSAV1E1Br | agggccctct agatgcatgc tcgagtcact cctcatcgct ggattcat | ||||
| CMVp-TPL | 2101SAV1E1Bs1 | gatagcggtt tgactcacgg | 293SE13 genome | 594 | |
| 2101SAV1E1Bs2 | gttctcctcc accactcggt | ||||
| SAV1E1B55K | 2101SAV1E1Bs3 | tggagcaaca gcgacagcc | 293SE13 genome | 811 | |
| 2101SAV1E1Bs4 | accgccttcc agcaaccat |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, X.; Sun, Y.; Chen, J.; Zou, X.; Hou, W.; Tan, W.; Hung, T.; Lu, Z. Restriction-Assembly: A Solution to Construct Novel Adenovirus Vector. Viruses 2022, 14, 546. https://doi.org/10.3390/v14030546
Guo X, Sun Y, Chen J, Zou X, Hou W, Tan W, Hung T, Lu Z. Restriction-Assembly: A Solution to Construct Novel Adenovirus Vector. Viruses. 2022; 14(3):546. https://doi.org/10.3390/v14030546
Chicago/Turabian StyleGuo, Xiaojuan, Yangyang Sun, Juan Chen, Xiaohui Zou, Wenzhe Hou, Wenjie Tan, Tao Hung, and Zhuozhuang Lu. 2022. "Restriction-Assembly: A Solution to Construct Novel Adenovirus Vector" Viruses 14, no. 3: 546. https://doi.org/10.3390/v14030546
APA StyleGuo, X., Sun, Y., Chen, J., Zou, X., Hou, W., Tan, W., Hung, T., & Lu, Z. (2022). Restriction-Assembly: A Solution to Construct Novel Adenovirus Vector. Viruses, 14(3), 546. https://doi.org/10.3390/v14030546