
Sequence Matching between Hemagglutinin and Neuraminidase through Sequence Analysis Using Machine Learning

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Preparation
2.2. Principal Component Analysis and Hierarchical Cluster Analysis
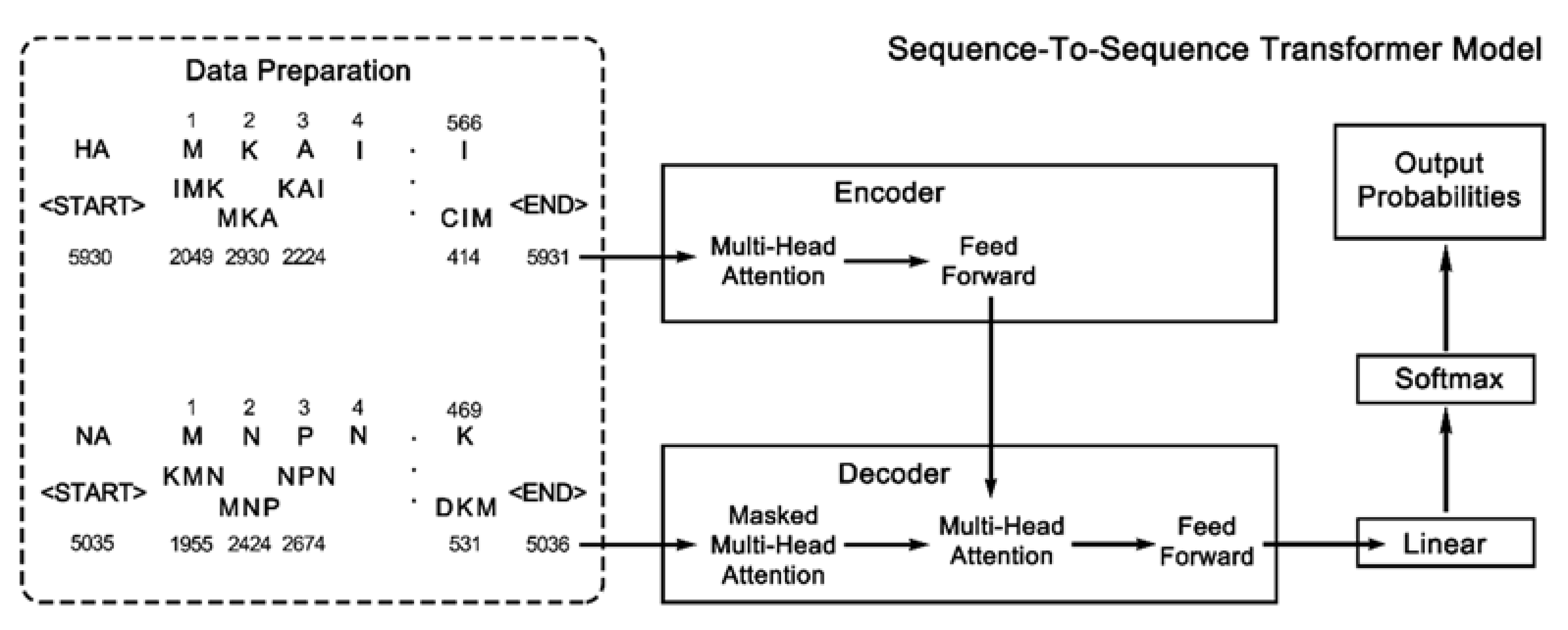
2.3. Sequence-to-Sequence Transformer Model
2.3.1. Word Set and Data Division
2.3.2. Multi-Head Attention Mechanism
2.3.3. Base Model
3. Results
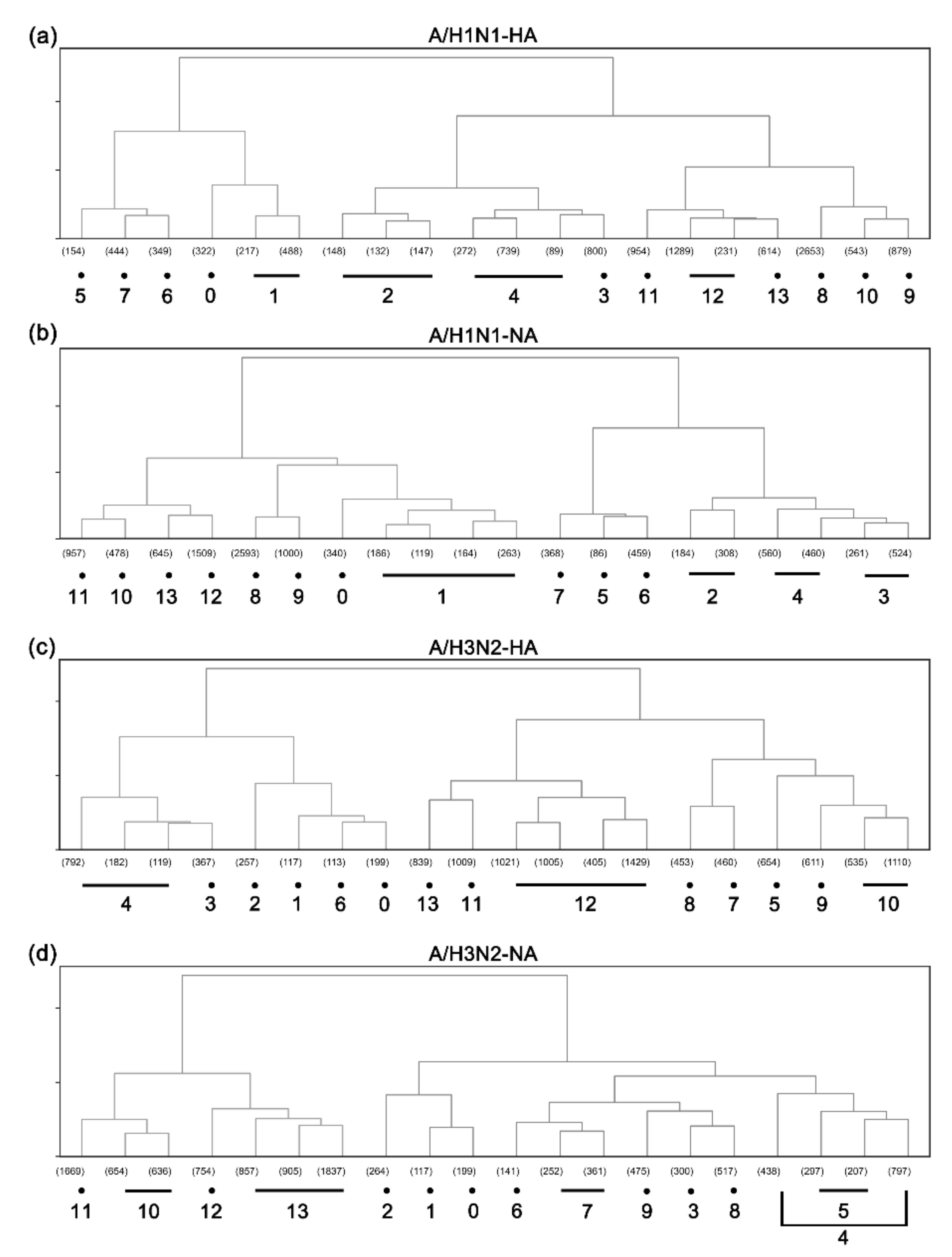
3.1. Hierarchical Clustering Analysis
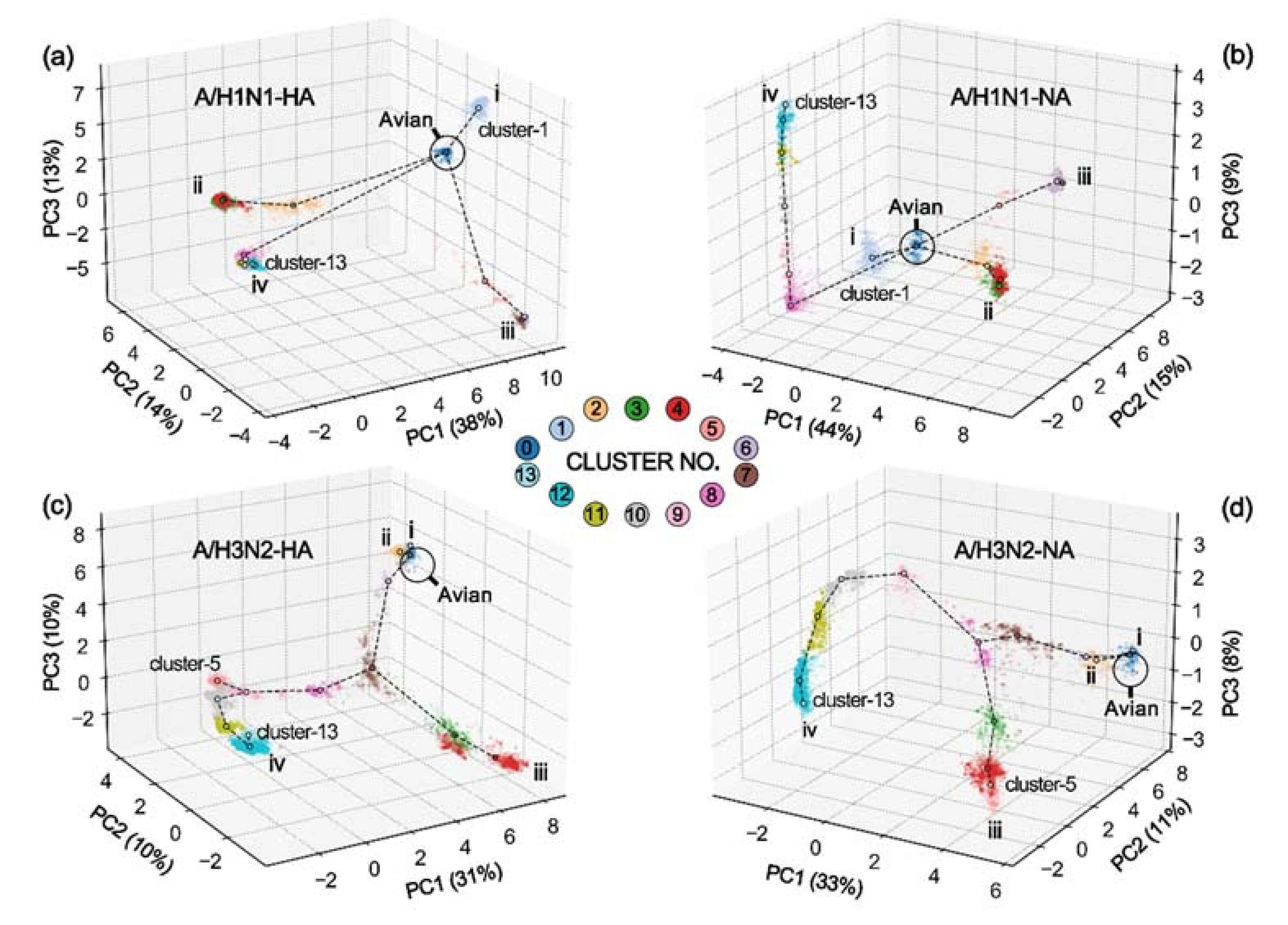
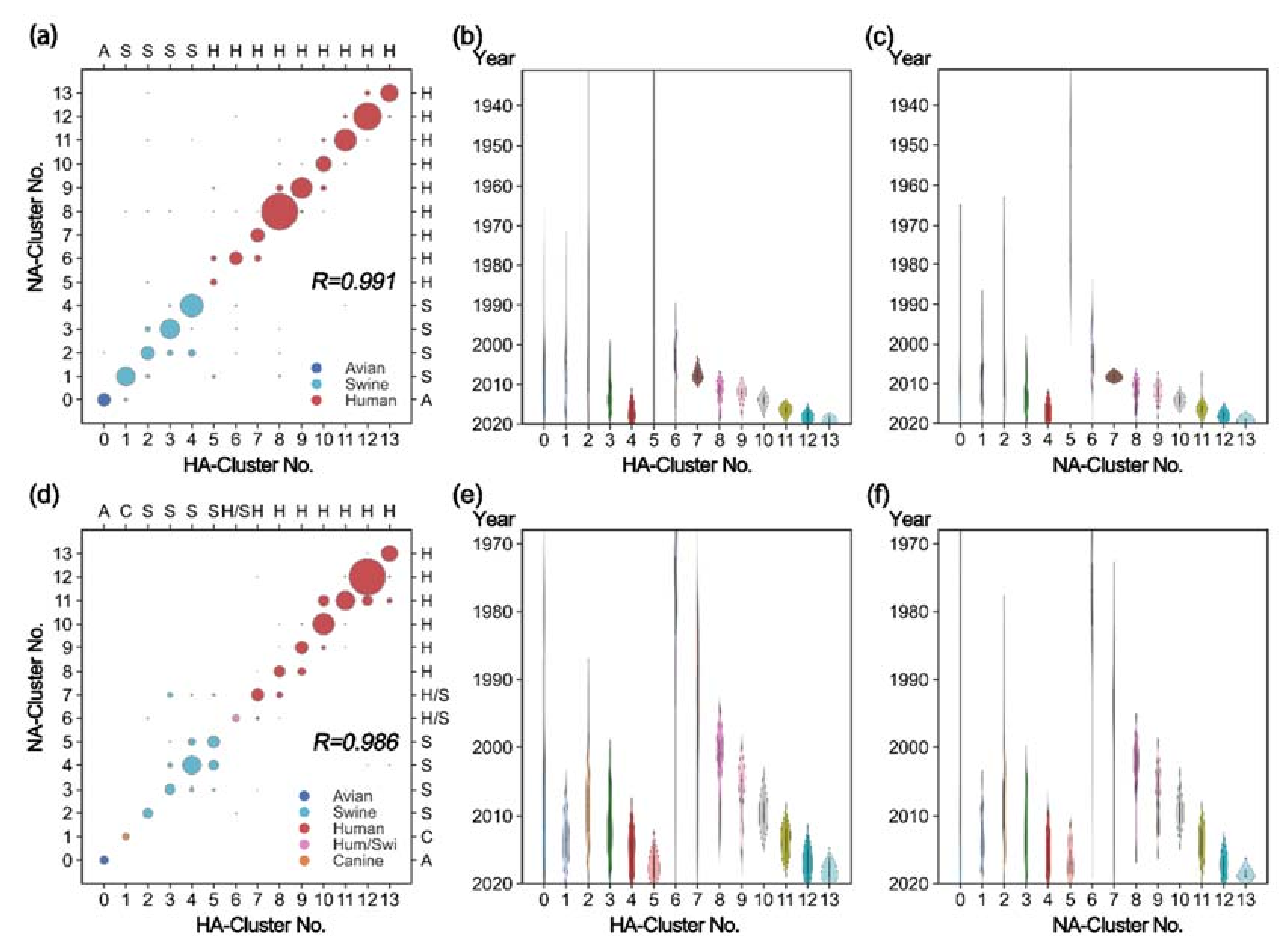
3.2. Coevolution between HA and NA
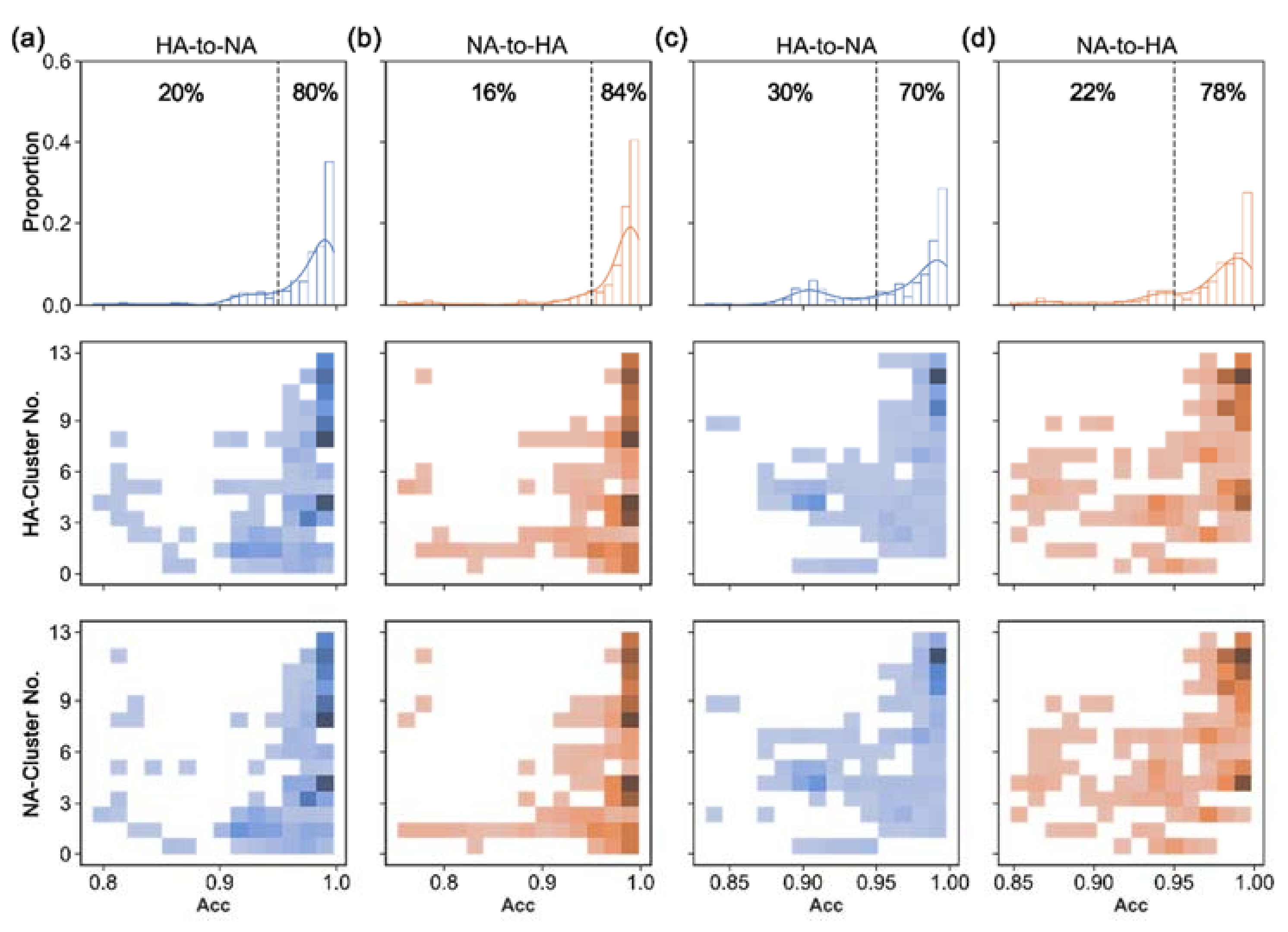
3.3. Translation Performance of S2STM
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wrapp, D.; Wang, N.S.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.B.; Holmes, E.C. Avian influenza virus exhibits rapid evolutionary dynamics. Mol. Biol. Evol. 2006, 23, 2336–2341. [Google Scholar] [CrossRef]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar] [CrossRef]
- Xu, X.J.; Zhu, X.Y.; Dwek, R.A.; Stevens, J.; Wilson, I.A. Structural Characterization of the 1918 Influenza Virus H1N1 Neuraminidase. J. Virol. 2008, 82, 10493–10501. [Google Scholar] [CrossRef]
- Gaymard, A.; Le Briand, N.; Frobert, E.; Lina, B.; Escuret, V. Functional balance between neuraminidase and haemagglutinin in influenza viruses. Clin. Microbiol. Infec. 2016, 22, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Kosik, I.; Yewdell, J.W. Influenza Hemagglutinin and Neuraminidase: Yin-Yang Proteins Coevolving to Thwart Immunity. Viruses-Basel 2019, 11, 346. [Google Scholar] [CrossRef]
- Medina, R.A.; Garcia-Sastre, A. Influenza A viruses: New research developments. Nat. Rev. Microbiol. 2011, 9, 590–603. [Google Scholar] [CrossRef]
- Olsen, C.W.; Karasin, A.I.; Carman, S.; Li, Y.; Bastien, N.; Ojkic, D.; Alves, D.; Charbonneau, G.; Henning, B.M.; Low, D.E.; et al. Triple reassortant H3N2 influenza A viruses, Canada, 2005. Emerg Infect Dis. 2006, 12, 1132–1135. [Google Scholar] [CrossRef]
- Schild, G.C.; Oxford, J.S.; Dowdle, W.R.; Coleman, M.T.; Chakraverty, P. Antigenic variation in current influenza A viruses. Evidence for a high frequency of antigenic ‘drift’ for the Hong Kong virus. Bull. World Health Organ. 1974, 51, 1–11. [Google Scholar]
- McAuley, J.L.; Gilbertson, B.P.; Trifkovic, S.; Brown, L.E.; McKimm-Breschkin, J.L. Influenza Virus Neuraminidase Structure and Functions. Front. Microbiol. 2019, 10, 39. [Google Scholar] [CrossRef]
- Byrd-Leotis, L.; Cummings, R.D.; Steinhauer, D.A. The Interplay between the Host Receptor and Influenza Virus Hemagglutinin and Neuraminidase. Int. J. Mol. Sci. 2017, 18, 1541. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Martin, S.R.; Wharton, S.A.; McCauley, J.W. Biophysical Measurement of the Balance of Influenza A Hemagglutinin and Neuraminidase Activities. J. Biol. Chem 2015, 290, 6516–6521. [Google Scholar] [CrossRef] [PubMed]
- de Vries, E.; Du, W.J.; Guo, H.B.; de Haan, C.A.M. Influenza A Virus Hemagglutinin-Neuraminidase-Receptor Balance: Preserving Virus Motility. Trends Microbiol. 2020, 28, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Rabouw, H.; Slomp, A.; Dai, M.; van der Vegt, F.; van Lent, J.W.M.; McBride, R.; Paulson, J.C.; de Groot, R.J.; van Kuppeveld, F.J.M.; et al. Kinetic analysis of the influenza A virus HA/NA balance reveals contribution of NA to virus-receptor binding and NA-dependent rolling on receptor-containing surfaces. PLoS Pathog. 2018, 14, e1007233. [Google Scholar] [CrossRef]
- Baigent, S.J.; McCauley, J.W. Glycosylation of haemagglutinin and stalk-length of neuraminidase combine to regulate the growth of avian influenza viruses in tissue culture. Virus Res. 2001, 79, 177–185. [Google Scholar] [CrossRef]
- Lai, J.C.C.; Karunarathna, H.M.T.K.; Wong, H.H.; Peiris, J.S.M.; Nicholls, J.M. Neuraminidase activity and specificity of influenza A virus are influenced by haemagglutinin-receptor binding. Emerg Microbes Infec. 2019, 8, 327–338. [Google Scholar] [CrossRef]
- Kaverin, N.V.; Gambaryan, A.S.; Bovin, N.V.; Rudneva, I.A.; Shilov, A.A.; Khodova, O.M.; Varich, N.L.; Sinitsin, B.V.; Makarova, N.V.; Kropotkina, E.A. Postreassortment changes in influenza A virus hemagglutinin restoring HA-NA functional match. Virology 1998, 244, 315–321. [Google Scholar] [CrossRef]
- Brown, I.H.; Ludwig, S.; Olsen, C.W.; Hannoun, C.; Scholtissek, C.; Hinshaw, V.S.; Harris, P.A.; McCauley, J.W.; Strong, I.; Alexander, D.J. Antigenic and genetic analyses of H1N1 influenza A viruses from European pigs. J. Gen. Virol. 1997, 78, 553–562. [Google Scholar] [CrossRef]
- Magge, A.; Weissenbacher, D.; Sarker, A.; Scotch, M.; Gonzalez-Hernandez, G. Deep neural networks and distant supervision for geographic location mention extraction. Bioinformatics 2018, 34, 565–573. [Google Scholar] [CrossRef]
- Bao, J.P.; Huang, F.F.; He, H.; Yang, K.; Feng, Z.H.; Wang, Y. Diversity and Evolution Trend of Protein Types of Human Influenza A (H1N1) Virus HA Segment. In Proceedings of the 2018 IEEE 20th International Conference on High Performance Computing and Communications; IEEE 16th International Conference on Smart City; IEEE 4th International Conference on Data Science and Systems (HPCC/SmartCity/DSS), Exeter, UK, 28–30 June 2018; pp. 1685–1691. [Google Scholar]
- Yin, R.; Luusua, E.; Dabrowski, J.; Zhang, Y.; Kwoh, C.K. Tempel: Time-series mutation prediction of influenza A viruses via attention-based recurrent neural networks. Bioinformatics 2020, 36, 2697–2704. [Google Scholar] [CrossRef]
- Ward, M.J.; Lycett, S.J.; Avila, D.; Bollback, J.P.; Brown, A.J.L. Evolutionary interactions between haemagglutinin and neuraminidase in avian influenza. BMC Evol. Biol. 2013, 13, 222. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.M.; Bolotov, P.; Dernovoy, D.; Kiryutin, B.; Zaslavsky, L.; Tatusova, T.; Ostell, J.; Lipman, D. The influenza virus resource at the national center for biotechnology information. J. Virol. 2008, 82, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tipping, M.E.; Bishop, C.M. Mixtures of probabilistic principal component analyzers. Neural Comput. 1999, 11, 443–482. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn Res. 2011, 12, 2825–2830. [Google Scholar]
- Buitinck, L.; Louppe, G.; Blondel, M.; Pedregosa, F.; Mueller, A.; Grisel, O.; Niculae, V.; Prettenhofer, P.; Gramfort, A.; Grobler, J.; et al. API design for machine learning software: Experiences from the scikit-learn project. arXiv 2013, arXiv:1309.0238. [Google Scholar]
- Bar-Joseph, Z.; Gifford, D.K.; Jaakkola, T.S. Fast optimal leaf ordering for hierarchical clustering. Bioinformatics 2001, 17 (Suppl. 1), S22–S29. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. arXiv 2017, arXiv:1706.03762. [Google Scholar]
- Abadi, M.; Barham, P.; Chen, J.M.; Chen, Z.F.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A system for large-scale machine learning. In Proceedings of the Osdi’16: 12th Usenix Symposium on Operating Systems Design and Implementation, Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Muller, M.; Lauster, D.; Wildenauer, H.H.K.; Herrmann, A.; Block, S. Mobility-Based Quantification of Multivalent Virus-Receptor Interactions: New Insights Into Influenza A Virus Binding Mode. Nano Lett. 2019, 19, 1875–1882. [Google Scholar] [CrossRef]
- Baigent, S.J.; Bethell, R.C.; McCauley, J.W. Genetic analysis reveals that both haemagglutinin and neuraminidase determine the sensitivity of naturally occurring avian influenza viruses to zanamivir in vitro. Virology 1999, 263, 323–338. [Google Scholar] [CrossRef]
- Wagner, R.; Matrosovich, M.; Klenk, H.D. Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev. Med. Virol. 2002, 12, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Tian, H.Z.; Nakaya, H.I. Antigenicity prediction and vaccine recommendation of human influenza virus A (H3N2) using convolutional neural networks. Hum. Vacc. Immunother. 2020, 16, 2690–2708. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.S.; Wang, D.Y.; Wang, J.H.; Li, K.L.; Tan, Z.Y.; Shu, Y.L.; Jiang, T.J. A universal computational model for predicting antigenic variants of influenza A virus based on conserved antigenic structures. Sci. Rep.-Uk 2017, 7, 42051. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.C.; Lee, M.S.; Ko, C.Y.; Hsiung, C.A. Bioinformatics models for predicting antigenic variants of influenza A/H3N2 virus. Bioinformatics 2008, 24, 505–512. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Branch | A/H1N1 | A/H3N2 | ||
|---|---|---|---|---|
| HA | NA | HA | NA | |
| i | 2.9 | 1.0 | 1.1 | 0.1 |
| ii | 10.7 | 4.5 | 0.9 | 0.6 |
| iii | 10.0 | 4.0 | 14.3 | 5.7 |
| iv | 11.8 | 6.0 | 22.5 | 8.6 |
| Division | A/H1N1 | A/H3N2 | ||
|---|---|---|---|---|
| HA-to-NA | NA-to-HA | HA-to-NA | NA-to-HA | |
| R1 | 0.9930 | 0.9923 | 0.9925 | 0.9927 |
| R2 | 0.9927 | 0.9920 | 0.9919 | 0.9929 |
| R3 | 0.9928 | 0.9918 | 0.9934 | 0.9928 |
| R4 | 0.9925 | 0.9918 | 0.9925 | 0.9926 |
| R5 | 0.9928 | 0.9918 | 0.9931 | 0.9927 |
| R6 | 0.9929 | 0.9921 | 0.9929 | 0.9925 |
| R7 | 0.9934 | 0.9921 | 0.9927 | 0.9927 |
| R8 | 0.9929 | 0.9919 | 0.9931 | 0.9929 |
| R9 | 0.9923 | 0.9918 | 0.9926 | 0.9927 |
| R10 | 0.9930 | 0.9921 | 0.9929 | 0.9929 |
| T | 0.9938 | 0.9931 | 0.9940 | 0.9938 |
| H* | 0.9954 | 0.9944 | 0.9950 | 0.9949 |
| Cluster | A/H1N1 | A/H3N2 | ||||||
|---|---|---|---|---|---|---|---|---|
| HA-to-NA | NA-to-HA | HA-to-NA | NA-to-HA | |||||
| Acc | Std | Acc | Std | Acc | Std | Acc | Std | |
| 0 | 0.89 | 0.008 | 0.82 | 0.004 | 0.85 | 0.014 | 0.84 | 0.009 |
| 1 | 0.91 | 0.010 | 0.78 | 0.008 | 0.83 | 0.016 | 0.83 | 0.006 |
| 2 | 0.83 | 0.009 | 0.90 | 0.015 | 0.85 | 0.011 | 0.84 | 0.006 |
| 3 | 0.83 | 0.004 | 0.92 | 0.007 | 0.94 | 0.021 | 0.88 | 0.013 |
| 4 | 0.82 | 0.003 | 0.91 | 0.007 | 0.93 | 0.019 | 0.87 | 0.013 |
| 5 | 0.83 | 0.012 | 0.80 | 0.010 | 0.90 | 0.007 | 0.93 | 0.010 |
| 6 | 0.82 | 0.004 | 0.79 | 0.003 | 0.90 | 0.018 | 0.86 | 0.018 |
| 7 | 0.81 | 0.002 | 0.79 | 0.003 | 0.95 | 0.021 | 0.90 | 0.014 |
| 13 | 0.98 | 0.004 | 0.98 | 0.011 | 0.96 | 0.003 | 0.96 | 0.015 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Zang, Y.; Zhao, Y.; Hao, D.; Kang, Y.; Zhang, J.; Zhang, Z.; Zhang, L.; Yang, Z.; Zhang, S. Sequence Matching between Hemagglutinin and Neuraminidase through Sequence Analysis Using Machine Learning. Viruses 2022, 14, 469. https://doi.org/10.3390/v14030469
Wang H, Zang Y, Zhao Y, Hao D, Kang Y, Zhang J, Zhang Z, Zhang L, Yang Z, Zhang S. Sequence Matching between Hemagglutinin and Neuraminidase through Sequence Analysis Using Machine Learning. Viruses. 2022; 14(3):469. https://doi.org/10.3390/v14030469
Chicago/Turabian StyleWang, He, Yongjian Zang, Yizhen Zhao, Dongxiao Hao, Ying Kang, Jianwen Zhang, Zichen Zhang, Lei Zhang, Zhiwei Yang, and Shengli Zhang. 2022. "Sequence Matching between Hemagglutinin and Neuraminidase through Sequence Analysis Using Machine Learning" Viruses 14, no. 3: 469. https://doi.org/10.3390/v14030469
APA StyleWang, H., Zang, Y., Zhao, Y., Hao, D., Kang, Y., Zhang, J., Zhang, Z., Zhang, L., Yang, Z., & Zhang, S. (2022). Sequence Matching between Hemagglutinin and Neuraminidase through Sequence Analysis Using Machine Learning. Viruses, 14(3), 469. https://doi.org/10.3390/v14030469