
V5 and GFP Tagging of Viral Gene pp38 of Marek’s Disease Vaccine Strain CVI988 Using CRISPR/Cas9 Editing



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viruses
2.2. Construction of sgRNA and Donor Template
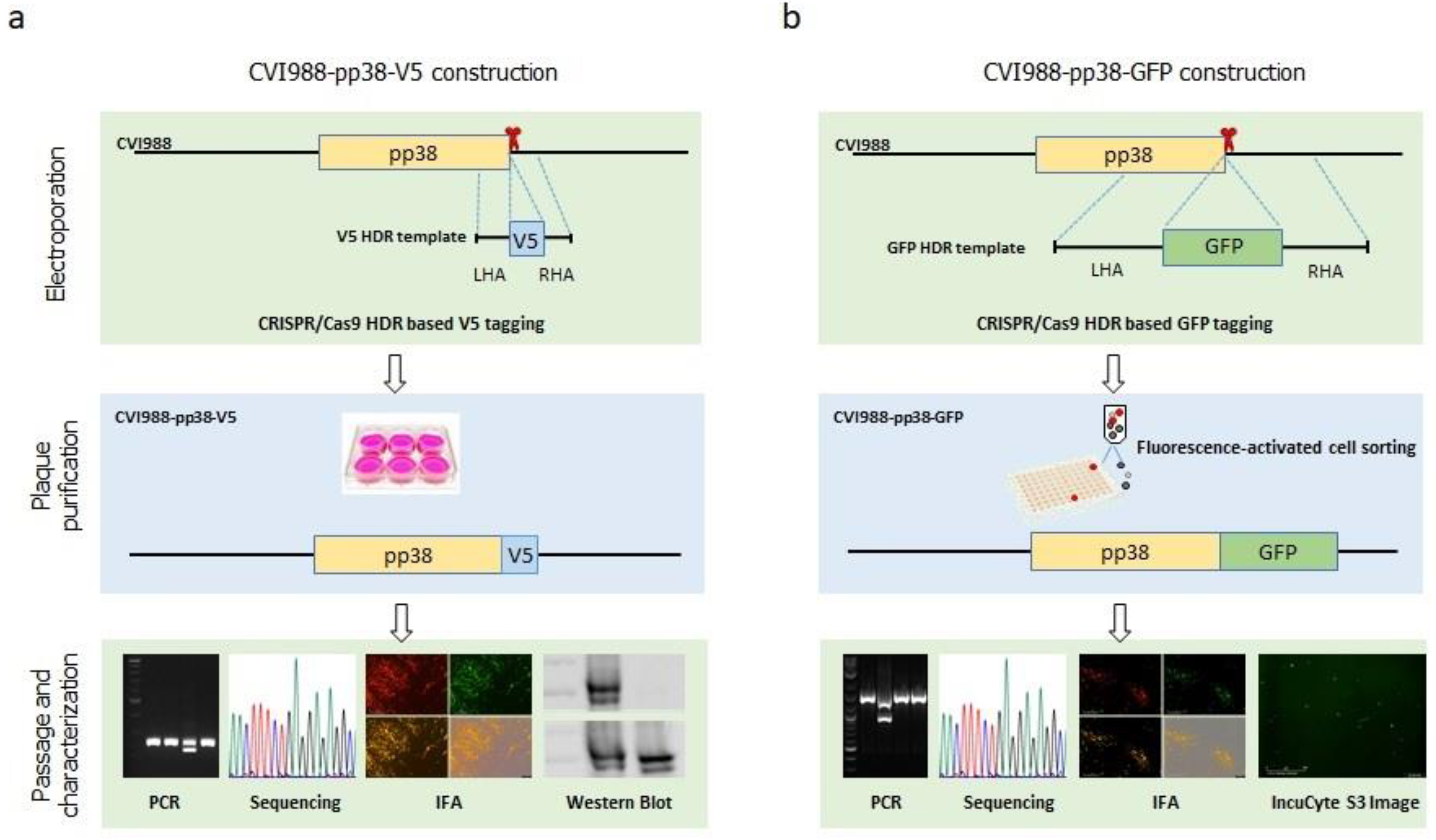
2.3. Generation and Characterization of V5- and GFP-Tagged CVI988 Viruses
2.4. Immunofluorescence Assay (IFA)
2.5. Single-Cell Sorting
2.6. Western Blotting
2.7. Live Cell Imaging of CVI988-pp38-GFP Growth
2.8. Stability of the Inserted Tags in the Recombinant Viruses
3. Results
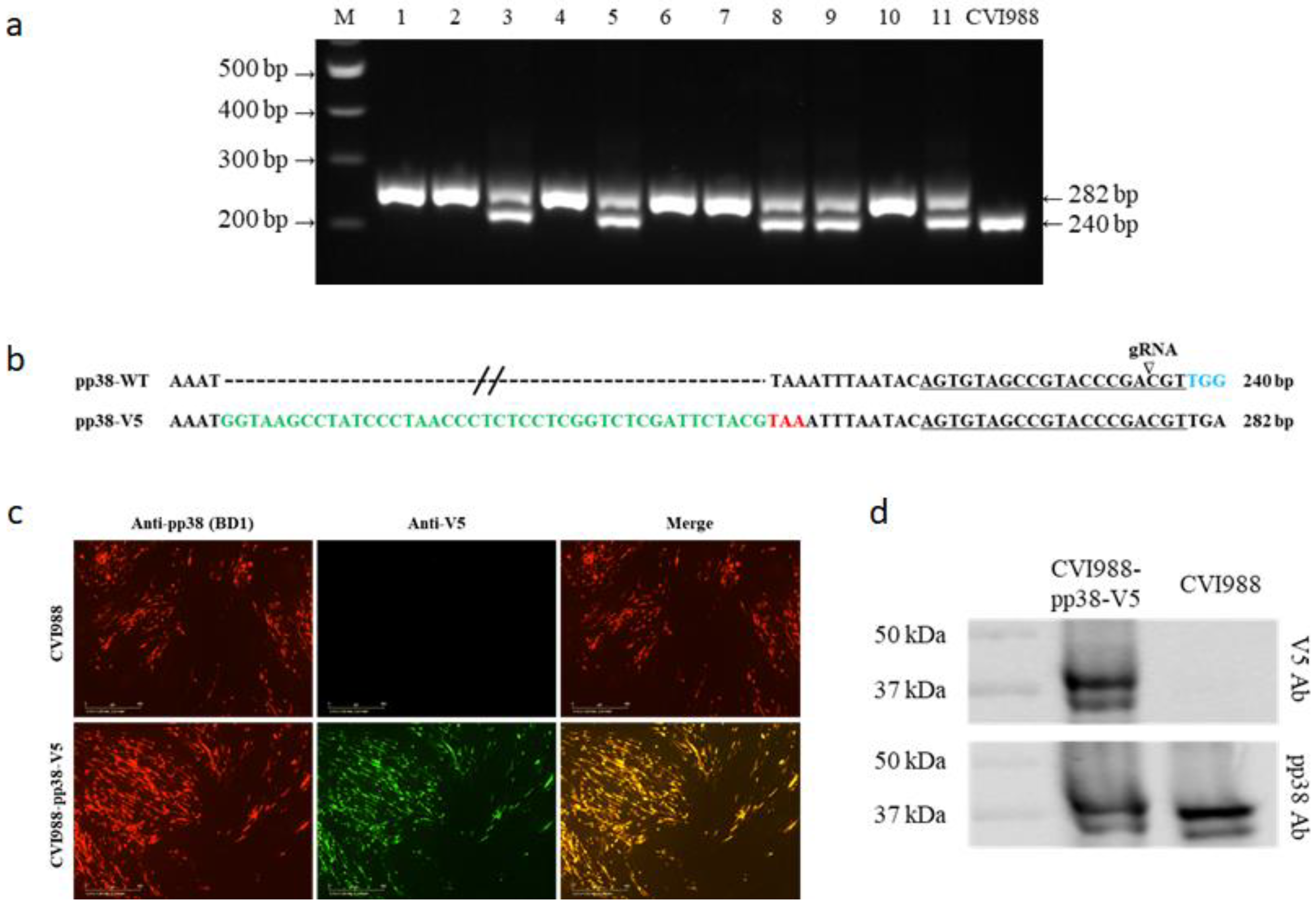
3.1. Generation of V5- and GFP-Tagged pp38 of CVI988 Virus Using CRISPR/Cas9 System
3.2. Characterization of CVI988-pp38-V5 and CVI988-pp38-GFP Viruses
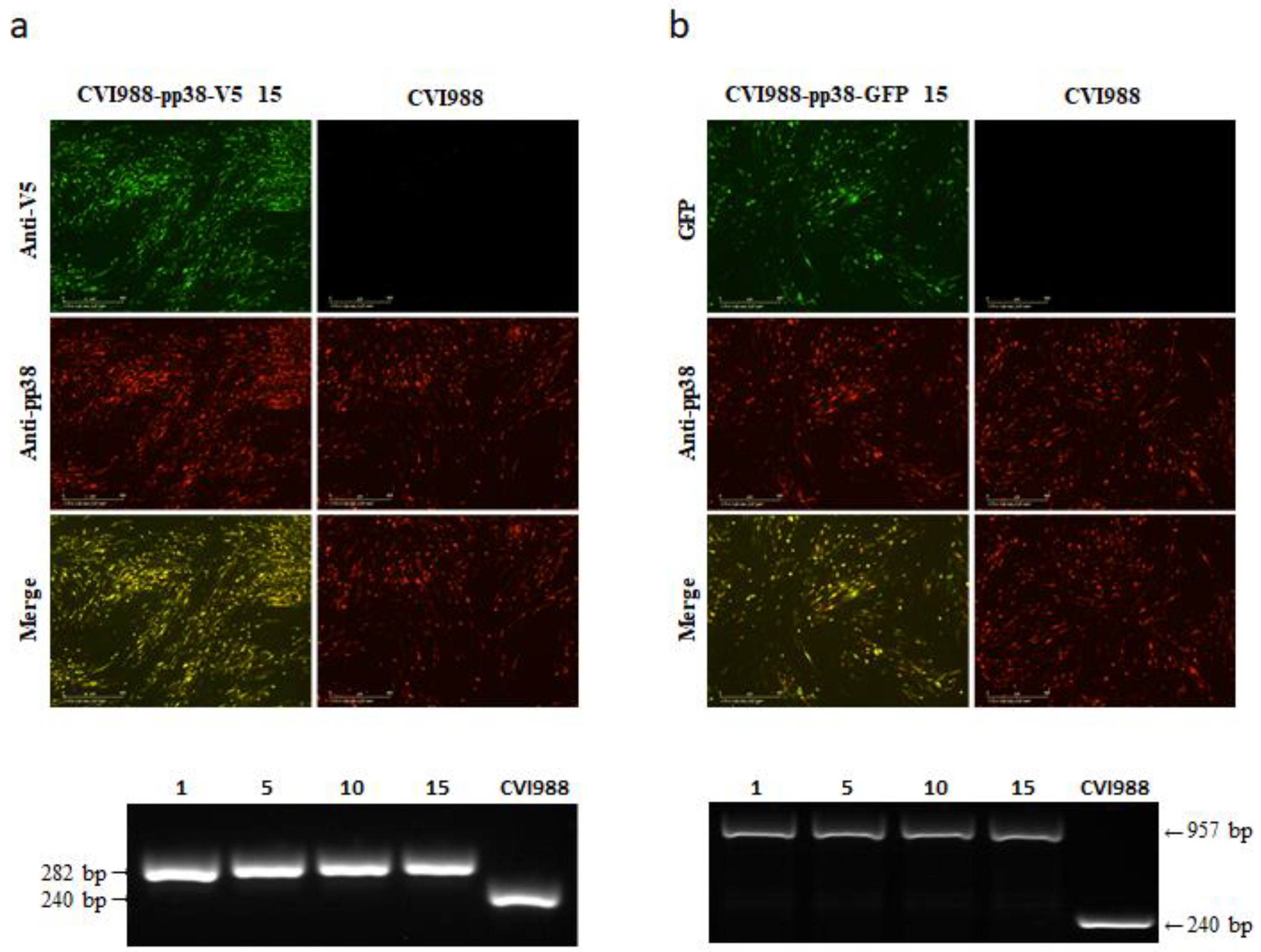
3.3. Stability of CVI988-pp38-V5 and CVI988-pp38-GFP
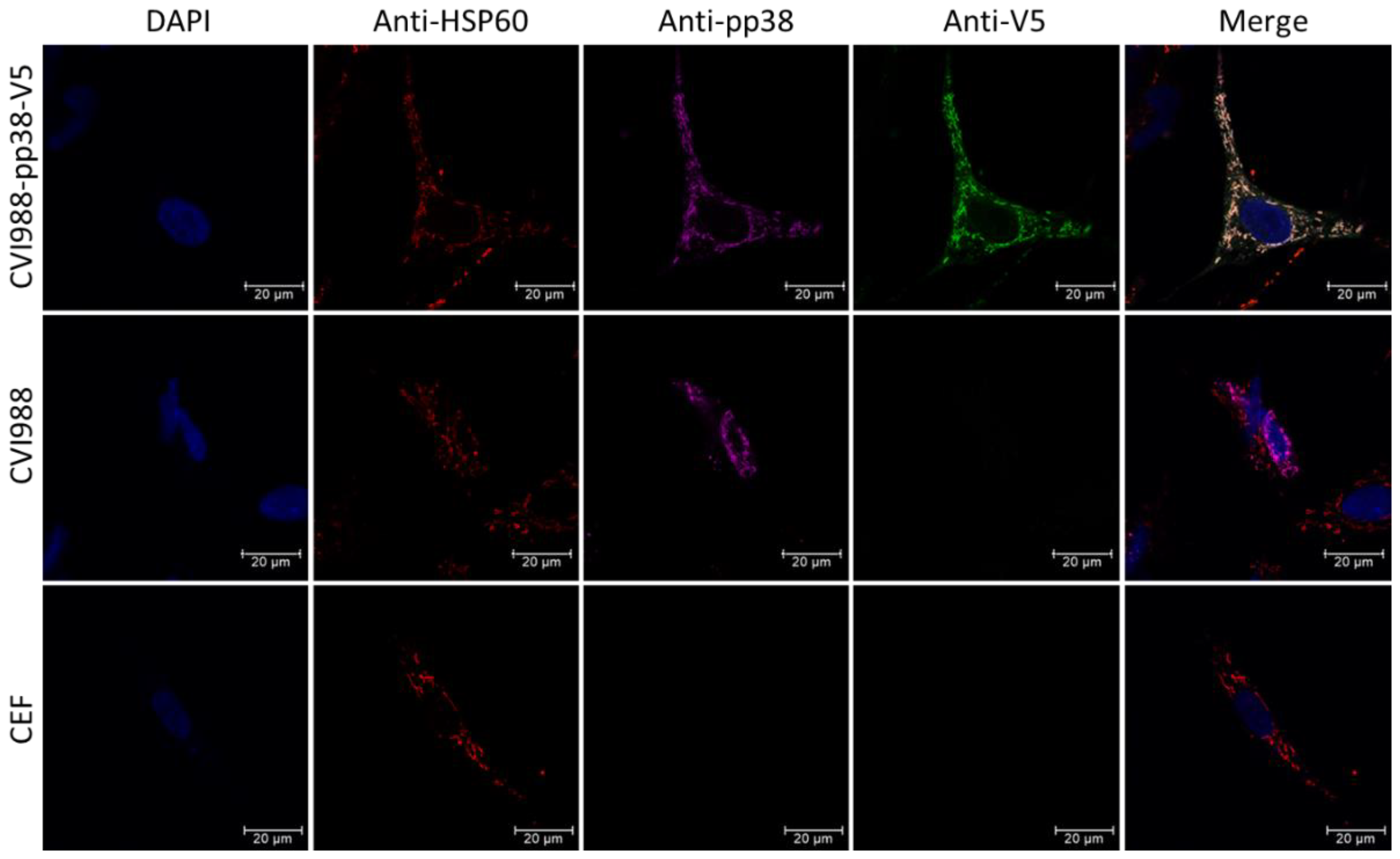
3.4. Possible Interaction of pp38 with Mitochondria Structure
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delecluse, H.J.; Hammerschmidt, W. Status of Marek’s disease virus in established lymphoma cell lines: Herpesvirus integration is common. J. Virol. 1993, 67, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Kaufer, B.B.; Jarosinski, K.W.; Osterrieder, N. Herpesvirus telomeric repeats facilitate genomic integration into host telomeres and mobilization of viral DNA during reactivation. J. Exp. Med. 2011, 208, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Delecluse, H.J.; Schuller, S.; Hammerschmidt, W. Latent Marek’s disease virus can be activated from its chromosomally integrated state in herpesvirus-transformed lymphoma cells. EMBO J. 1993, 12, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Biggs, P.M. The Leeuwenhoek Lecture, 1997. Marek’s disease herpesvirus: Oncogenesis and prevention. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1997, 352, 1951–1962. [Google Scholar] [CrossRef]
- Venugopal, K. Marek’s disease: An update on oncogenic mechanisms and control. Res. Vet. Sci. 2000, 69, 17–23. [Google Scholar] [CrossRef]
- Lee, L.F.; Lupiani, B.; Silva, R.F.; Kung, H.J.; Reddy, S.M. Recombinant Marek’s disease virus (MDV) lacking the Meq oncogene confers protection against challenge with a very virulent plus strain of MDV. Vaccine 2008, 26, 1887–1892. [Google Scholar] [CrossRef]
- Silva, R.F.; Dunn, J.R.; Cheng, H.H.; Niikura, M. A MEQ-Deleted Marek’s Disease Virus Cloned as a Bacterial Artificial Chromosome Is a Highly Efficacious Vaccine. Avian Dis. 2010, 54, 862–869. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Liu, C.J.; Yan, F.H.; Liu, A.L.; Cheng, Y.; Li, Z.J.; Sun, G.R.; Lv, H.C.; Wang, X.M. Recombinant Gallid herpesvirus 2 with interrupted meq genes confers safe and efficacious protection against virulent field strains. Vaccine 2017, 35, 4695–4701. [Google Scholar] [CrossRef]
- Lupiani, B.; Lee, L.F.; Cui, X.P.; Gimeno, I.; Anderson, A.; Morgan, R.W.; Silva, R.F.; Witter, R.L.; Kung, H.J.; Reddy, S.M. Marek’s disease virus-encoded Meq gene is involved in transformation of lymphocytes but is dispensable for replication. Proc. Natl. Acad. Sci. USA 2004, 101, 11815–11820. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Xu, H.T.; Yao, Y.X.; Smith, L.P.; Kgosana, L.; Green, J.; Petherbridge, L.; Baigent, S.J.; Nair, V. Critical Role of the Virus-Encoded MicroRNA-155 Ortholog in the Induction of Marek’s Disease Lymphomas. PLoS Pathogens 2011, 7, e1001305. [Google Scholar] [CrossRef]
- Trapp, S.; Parcells, M.S.; Kamil, J.P.; Schumacher, D.; Tischer, B.K.; Kumar, P.M.; Nair, V.K.; Osterrieder, N. A virus-encoded telomerase RNA promotes malignant T cell lymphomagenesis. J. Exp. Med. 2006, 203, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Fester, N.; Engel, A.T.; Kaufer, B.B. Role of the Short Telomeric Repeat Region in Marek’s Disease Virus Replication, Genomic Integration, and Lymphomagenesis. J. Virol. 2014, 88, 14138–14147. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Baigent, S.J.; Smith, L.P.; Chattoo, J.P.; Petherbridge, L.J.; Hawes, P.; Allday, M.J.; Nair, V. Interaction of MEQ protein and C-terminal-binding protein is critical for induction of lymphomas by Marek’s disease virus. Proc. Natl. Acad. Sci. USA 2006, 103, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, A.; Su, S.; Zhao, P.; Cui, Z.; Zhu, H. Deletion of the Meq gene significantly decreases immunosuppression in chickens caused by pathogenic Marek’s disease virus. Virol. J. 2011, 8, 2. [Google Scholar] [CrossRef]
- Cui, Z.Z.; Lee, L.F.; Liu, J.L.; Kung, H.J. Structural analysis and transcriptional mapping of the Marek’s disease virus gene encoding pp38, an antigen associated with transformed cells. J. Virol. 1991, 65, 6509–6515. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.M.; Lupiani, B.; Gimeno, I.M.; Silva, R.F.; Lee, L.F.; Witter, R.L. Rescue of a pathogenic Marek’s disease virus with overlapping cosmid DNAs: Use of a pp38 mutant to validate the technology for the study of gene function. Proc. Natl. Acad. Sci. USA 2002, 99, 7054–7059. [Google Scholar] [CrossRef] [PubMed]
- Fragnet, L.; Blasco, M.A.; Klapper, W.; Rasschaert, D. The RNA subunit of telomerase is encoded by Marek’s disease virus. J. Virol. 2003, 77, 5985–5996. [Google Scholar] [CrossRef]
- Cortes, P.L.; Cardona, C.J. Pathogenesis of a Marek’s disease virus mutant lacking vIL-8 in resistant and susceptible chickens. Avian Dis. 2004, 48, 50–60. [Google Scholar] [CrossRef]
- Cui, X.; Lee, L.F.; Hunt, H.D.; Reed, W.M.; Lupiani, B.; Reddy, S.M. A Marek’s disease virus vIL-8 deletion mutant has attenuated virulence and confers protection against challenge with a very virulent plus strain. Avian Dis. 2005, 49, 199–206. [Google Scholar] [CrossRef]
- Cui, X.; Lee, L.F.; Reed, W.M.; Kung, H.J.; Reddy, S.M. Marek’s disease virus-encoded vIL-8 gene is involved in early cytolytic infection but dispensable for establishment of latency. J. Virol. 2004, 78, 4753–4760. [Google Scholar] [CrossRef]
- Parcells, M.S.; Lin, S.F.; Dienglewicz, R.L.; Majerciak, V.; Robinson, D.R.; Chen, H.C.; Wu, Z.; Dubyak, G.R.; Brunovskis, P.; Hunt, H.D.; et al. Marek’s disease virus (MDV) encodes an interleukin-8 homolog (vIL-8): Characterization of the vIL-8 protein and a vIL-8 deletion mutant MDV. J. Virol. 2001, 75, 5159–5173. [Google Scholar] [CrossRef] [PubMed]
- Jarosinski, K.W.; Osterrieder, N.; Nair, V.K.; Schat, K.A. Attenuation of Marek’s disease virus by deletion of open reading frame RLORF4 but not RLORF5a. J. Virol. 2005, 79, 11647–11659. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Lee, L.F.; Khan, O.A.; Heidari, M.; Zhang, H.; Lupiani, B.; Reddy, S.M. Deletion of Marek’s disease virus large subunit of ribonucleotide reductase impairs virus growth in vitro and in vivo. Avian Dis. 2013, 57, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Dorange, F.; Tischer, B.K.; Vautherot, J.F.; Osterrieder, N. Characterization of Marek’s disease virus serotype 1 (MDV-1) deletion mutants that lack UL46 to UL49 genes: MDV-1 UL49, encoding VP22, is indispensable for virus growth. J. Virol. 2002, 76, 1959–1970. [Google Scholar] [CrossRef]
- van der Oost, J.; Jore, M.M.; Westra, E.R.; Lundgren, M.; Brouns, S.J.J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 2009, 34, 401–407. [Google Scholar] [CrossRef]
- Xu, A.T.; Qin, C.; Lang, Y.; Wang, M.Y.; Lin, M.Y.; Li, C.; Zhang, R.; Tang, J. A simple and rapid approach to manipulate pseudorabies virus genome by CRISPR/Cas9 system. Biotechnol. Lett. 2015, 37, 1265–1272. [Google Scholar] [CrossRef]
- Liang, X.; Sun, L.Q.; Yu, T.; Pan, Y.F.; Wang, D.D.; Hu, X.Y.; Fu, Z.F.; He, Q.G.; Cao, G. A CRISPR/Cas9 and Cre/Lox system-based express vaccine development strategy against re-emerging Pseudorabies virus. Sci. Rep. 2016, 6, 19176. [Google Scholar] [CrossRef]
- Peng, Z.Y.; Ouyang, T.; Pang, D.X.; Ma, T.; Chen, X.R.; Guo, N.; Chen, F.W.; Yuan, L.; Ouyang, H.S.; Ren, L.Z. Pseudorabies virus can escape from CRISPR-Cas9-mediated inhibition. Virus Res. 2016, 223, 197–205. [Google Scholar] [CrossRef]
- Tang, Y.D.; Liu, J.T.; Wang, T.Y.; An, T.Q.; Sun, M.X.; Wang, S.J.; Fang, Q.Q.; Hou, L.L.; Tian, Z.J.; Cai, X.H. Live attenuated pseudorabies virus developed using the CRISPR/Cas9 system. Virus Res. 2016, 225, 33–39. [Google Scholar] [CrossRef]
- Yuan, M.; Zhang, W.S.; Wang, J.; Al Yaghchi, C.; Ahmed, J.; Chard, L.; Lemoine, N.R.; Wang, Y.H. Efficiently Editing the Vaccinia Virus Genome by Using the CRISPR-Cas9 System. J. Virol. 2015, 89, 5176–5179. [Google Scholar] [CrossRef]
- Tso, F.Y.; West, J.T.; Wood, C. Reduction of Kaposi’s Sarcoma-Associated Herpesvirus Latency Using CRISPR-Cas9 To Edit the Latency-Associated Nuclear Antigen Gene. J. Virol. 2019, 93, e02183-18. [Google Scholar] [CrossRef]
- Gergen, J.; Coulon, F.; Creneguy, A.; Elain-Duret, N.; Gutierrez, A.; Pinkenburg, O.; Verhoeyen, E.; Anegon, I.; Nguyen, T.H.; Halary, F.A.; et al. Multiplex CRISPR/Cas9 system impairs HCMV replication by excising an essential viral gene. PLoS ONE 2018, 13, e0192602. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Deng, J.; Zhang, Q.; Ma, P.; Lv, L.; Zhang, Y.; Li, C.; Zhang, Y. Targeting human cytomegalovirus IE genes by CRISPR/Cas9 nuclease effectively inhibits viral replication and reactivation. Arch. Virol. 2020, 165, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.S.; Chan, C.P.; Wong, N.H.M.; Ho, C.H.; Ho, T.H.; Lei, T.; Deng, W.; Tsao, S.W.; Chen, H.L.; Kok, K.H.; et al. CRISPR/Cas9-mediated genome editing of Epstein Barr virus in human cells. J. Gen. Virol. 2015, 96, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Bierle, C.J.; Anderholm, K.M.; Ben Wang, J.; McVoy, M.A.; Schleiss, M.R. Targeted Mutagenesis of Guinea Pig Cytomegalovirus Using CRISPR/Cas9-Mediated Gene Editing. J. Virol. 2016, 90, 6989–6998. [Google Scholar] [CrossRef]
- Zou, Z.; Huang, K.; Wei, Y.M.; Chen, H.C.; Liu, Z.D.; Jin, M.L. Construction of a highly efficient CRISPR/Cas9-mediated duck enteritis virus-based vaccine against H5N1 avian influenza virus and duck Tembusu virus infection. Sci. Rep. 2017, 7, 1478. [Google Scholar] [CrossRef]
- Tang, N.; Zhang, Y.; Pedrera, M.; Chang, P.; Baigent, S.; Moffat, K.; Shen, Z.; Nair, V.; Yao, Y. A simple and rapid approach to develop recombinant avian herpesvirus vectored vaccines using CRISPR/Cas9 system. Vaccine 2018, 36, 716–722. [Google Scholar] [CrossRef]
- Tang, N.; Zhang, Y.; Sadigh, Y.; Moffat, K.; Shen, Z.; Nair, V.; Yao, Y. Generation of A Triple Insert Live Avian Herpesvirus Vectored Vaccine Using CRISPR/Cas9-Based Gene Editing. Vaccines 2020, 8, 97. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, J.; Tang, N.; Teng, M.; Reddy, V.; Moffat, K.; Shen, Z.; Nair, V.; Yao, Y. Targeted Editing of the pp38 Gene in Marek’s Disease Virus-Transformed Cell Lines Using CRISPR/Cas9 System. Viruses 2019, 11, 391. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, N.; Luo, J.; Teng, M.; Moffat, K.; Shen, Z.; Watson, M.; Nair, V.; Yao, Y. Marek’s Disease Virus-Encoded MicroRNA 155 Ortholog Critical for the Induction of Lymphomas Is Not Essential for the Proliferation of Transformed Cell Lines. J. Virol. 2019, 93, e00713-19. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, N.; Sadigh, Y.; Baigent, S.; Shen, Z.; Nair, V.; Yao, Y. Application of CRISPR/Cas9 Gene Editing System on MDV-1 Genome for the Study of Gene Function. Viruses 2018, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Teng, M.; Zai, X.; Tang, N.; Zhang, Y.; Mandviwala, A.; Reddy, V.; Baigent, S.; Yao, Y.; Nair, V. Efficient Mutagenesis of Marek’s Disease Virus-Encoded microRNAs Using a CRISPR/Cas9-Based Gene Editing System. Viruses 2020, 12, 466. [Google Scholar] [CrossRef] [PubMed]
- Hagag, I.T.; Wight, D.J.; Bartsch, D.; Sid, H.; Jordan, I.; Bertzbach, L.D.; Schusser, B.; Kaufer, B.B. Abrogation of Marek’s disease virus replication using CRISPR/Cas9. Sci. Rep. 2020, 10, 10919. [Google Scholar] [CrossRef] [PubMed]
- Challagulla, A.; Jenkins, K.A.; O’Neil, T.E.; Shi, S.; Morris, K.R.; Wise, T.G.; Paradkar, P.N.; Tizard, M.L.; Doran, T.J.; Schat, K.A. In Vivo Inhibition of Marek’s Disease Virus in Transgenic Chickens Expressing Cas9 and gRNA against ICP4. Microorganisms 2021, 9, 164. [Google Scholar] [CrossRef]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, S.; Hur, J.K. CRISPR and Target-Specific DNA Endonucleases for Efficient DNA Knock-in in Eukaryotic Genomes. Mol. Cells. 2018, 41, 943–952. [Google Scholar] [CrossRef]
- Gimeno, I.M.; Witter, R.L.; Hunt, H.D.; Reddy, S.M.; Lee, L.F.; Silva, R.F. The pp38 gene of Marek’s disease virus (MDV) is necessary for cytolytic infection of B cells and maintenance of the transformed state but not for cytolytic infection of the feather follicle epithelium and horizontal spread of MDV. J. Virol. 2005, 79, 4545–4549. [Google Scholar] [CrossRef]
- Lupiani, B.M.; Liao, Y.; Jin, D.; Izumiya, Y.; Reddy, S.M. Marek’s Disease Virus. In Avian Virology: Current Research and Future Trends; Caister Academic Press: Poole, UK, 2019; pp. 345–364. [Google Scholar] [CrossRef]
- Roy, P.; Moffat, K.; Nair, V.; Yao, Y. CRISPR-Mediated Gene Activation (CRISPRa) of pp38/pp24 Orchestrates Events Triggering Lytic Infection in Marek’s Disease Virus-Transformed Cell Lines. Microorganisms 2021, 9, 1681. [Google Scholar] [CrossRef]
- Yao, Y.; Bassett, A.; Nair, V. Targeted editing of avian herpesvirus vaccine vector using CRISPR/Cas9 nucleases. J. Vaccines Technol. 2016, 1, 1–7. [Google Scholar]
- Reddy, V.; Sadigh, Y.; Tang, N.; Yao, Y.; Nair, V. Novel Insights into the Roles of Bcl-2 Homolog Nr-13 (vNr-13) Encoded by Herpesvirus of Turkeys in the Virus Replication Cycle, Mitochondrial Networks, and Apoptosis Inhibition. J. Virol. 2020, 94, e02049-19. [Google Scholar] [CrossRef]
- Tang, N.; Zhang, Y.; Pedrera, M.; Chang, P.; Baigent, S.; Moffat, K.; Shen, Z.; Nair, V.; Yao, Y. Generating Recombinant Avian Herpesvirus Vectors with CRISPR/Cas9 Gene Editing. J. Vis. Exp. 2019, 143, e58193. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef]
- Irion, U.; Krauss, J.; Nusslein-Volhard, C. Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development 2014, 141, 4827–4830. [Google Scholar] [CrossRef]
- Hisano, Y.; Sakuma, T.; Nakade, S.; Ohga, R.; Ota, S.; Okamoto, H.; Yamamoto, T.; Kawahara, A. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Sci. Rep. 2015, 5, 8841. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, K.Y.; Jung, K.M.; Park, K.J.; Lee, K.O.; Suh, J.Y.; Yao, Y.; Nair, V.; Han, J.Y. Precise gene editing of chicken Na+/H+ exchange type 1 (chNHE1) confers resistance to avian leukosis virus subgroup J (ALV-J). Dev. Comp. Immunol. 2017, 77, 340–349. [Google Scholar] [CrossRef]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Kaplan, S.L.; Wakenell, P.; Schat, K.A. Transactivation of latent Marek’s disease herpesvirus genes in QT35, a quail fibroblast cell line, by herpesvirus of turkeys. J. Virol. 2000, 74, 10176–10186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, X.; Jarosinski, K.W.; Schat, K.A. Expression of Marek’s disease virus phosphorylated polypeptide pp38 produces splice variants and enhances metabolic activity. Vet. Microbiol. 2006, 117, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Piepenbrink, M.S.; Li, X.; O’Connell, P.H.; Schat, K.A. Marek’s disease virus phosphorylated polypeptide pp38 alters transcription rates of mitochondrial electron transport and oxidative phosphorylation genes. Virus Genes 2009, 39, 102–112. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′–3′) |
|---|---|
| pp38-gRNA-F | CACCGGTGTAGCCGTACCCGACGT |
| pp38-gRNA-R | AAACACGTCGGGTACGGCTACACC |
| pp38-tag-F | GTTGGGGGAGTGGATTCTGG |
| pp38-tag-R | TCCTCGATTGGTGGGGAGAT |
| pp38-GFP-A-F | CACCGCGACCCGAGAGAAAGATCG |
| pp38-GFP-A-R | TCCTCGCCCTTGCTCACCATATTTGATTCAGATTTTGTTT |
| pp38-GFP-B-F | AAACAAAATCTGAATCAAATATGGTGAGCAAGGGCGAGGA |
| pp38-GFP-B-R | CTCCGCCTTCAACGTCGGGTACGGCTACACTGTATTAAATTTACTTGTACAGCTCGTCCA |
| pp38-GFP-C-F | TGGACGAGCTGTACAAGTAAATTTAATACAGTGTAGCCGTACCCGACGTTGAAGGCGGAG |
| pp38-GFP-C-R | CATCCGAGTGGTACGTGCAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Zhang, Y.; Moffat, K.; Nair, V.; Yao, Y. V5 and GFP Tagging of Viral Gene pp38 of Marek’s Disease Vaccine Strain CVI988 Using CRISPR/Cas9 Editing. Viruses 2022, 14, 436. https://doi.org/10.3390/v14020436
Li W, Zhang Y, Moffat K, Nair V, Yao Y. V5 and GFP Tagging of Viral Gene pp38 of Marek’s Disease Vaccine Strain CVI988 Using CRISPR/Cas9 Editing. Viruses. 2022; 14(2):436. https://doi.org/10.3390/v14020436
Chicago/Turabian StyleLi, Weicheng, Yaoyao Zhang, Katy Moffat, Venugopal Nair, and Yongxiu Yao. 2022. "V5 and GFP Tagging of Viral Gene pp38 of Marek’s Disease Vaccine Strain CVI988 Using CRISPR/Cas9 Editing" Viruses 14, no. 2: 436. https://doi.org/10.3390/v14020436
APA StyleLi, W., Zhang, Y., Moffat, K., Nair, V., & Yao, Y. (2022). V5 and GFP Tagging of Viral Gene pp38 of Marek’s Disease Vaccine Strain CVI988 Using CRISPR/Cas9 Editing. Viruses, 14(2), 436. https://doi.org/10.3390/v14020436