
Comparative Investigation of Methods for Analysis of SARS-CoV-2-Spike-Specific Antisera

 , , , , , , ,
, , , , , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Protein Production and Purification
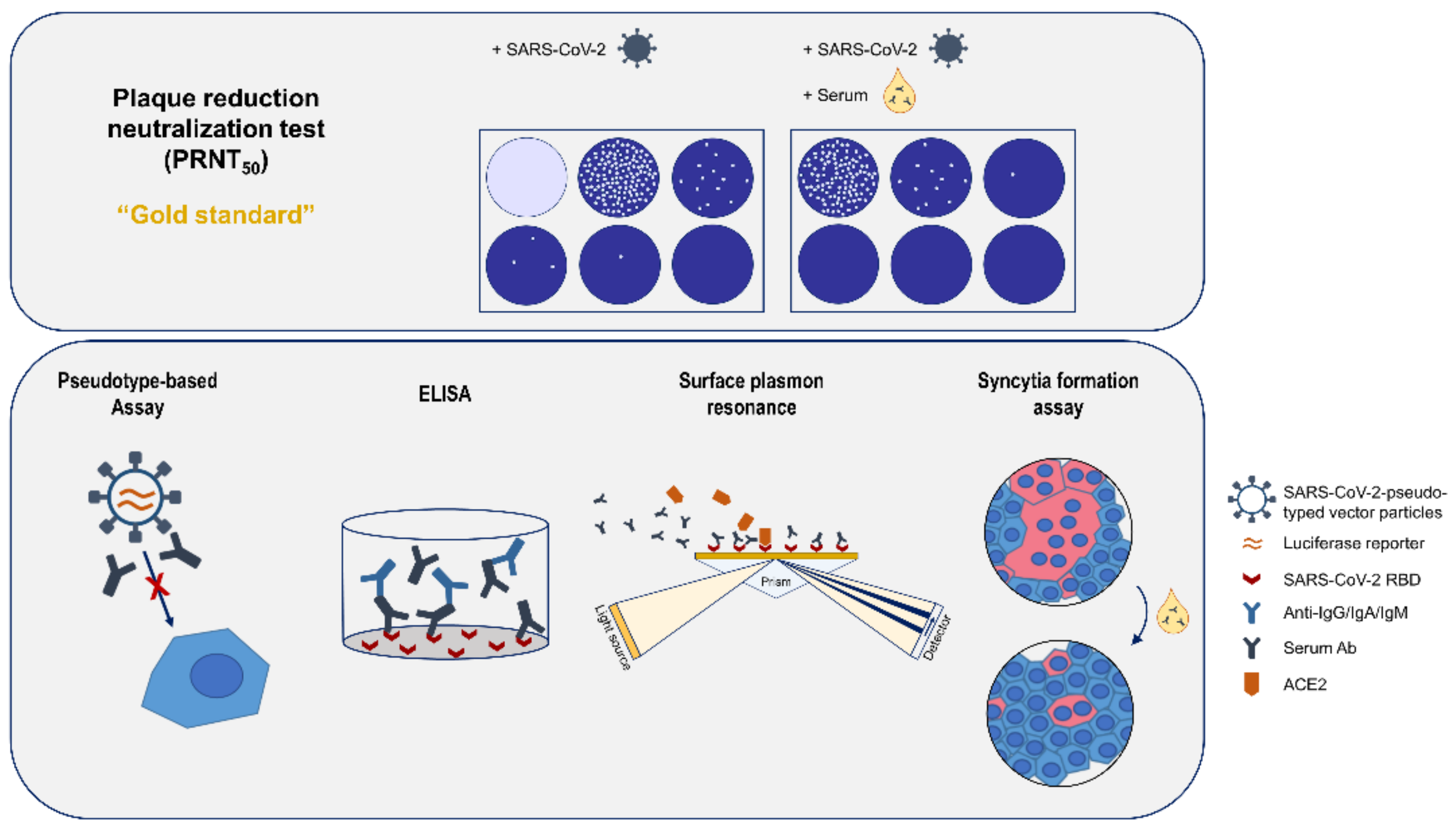
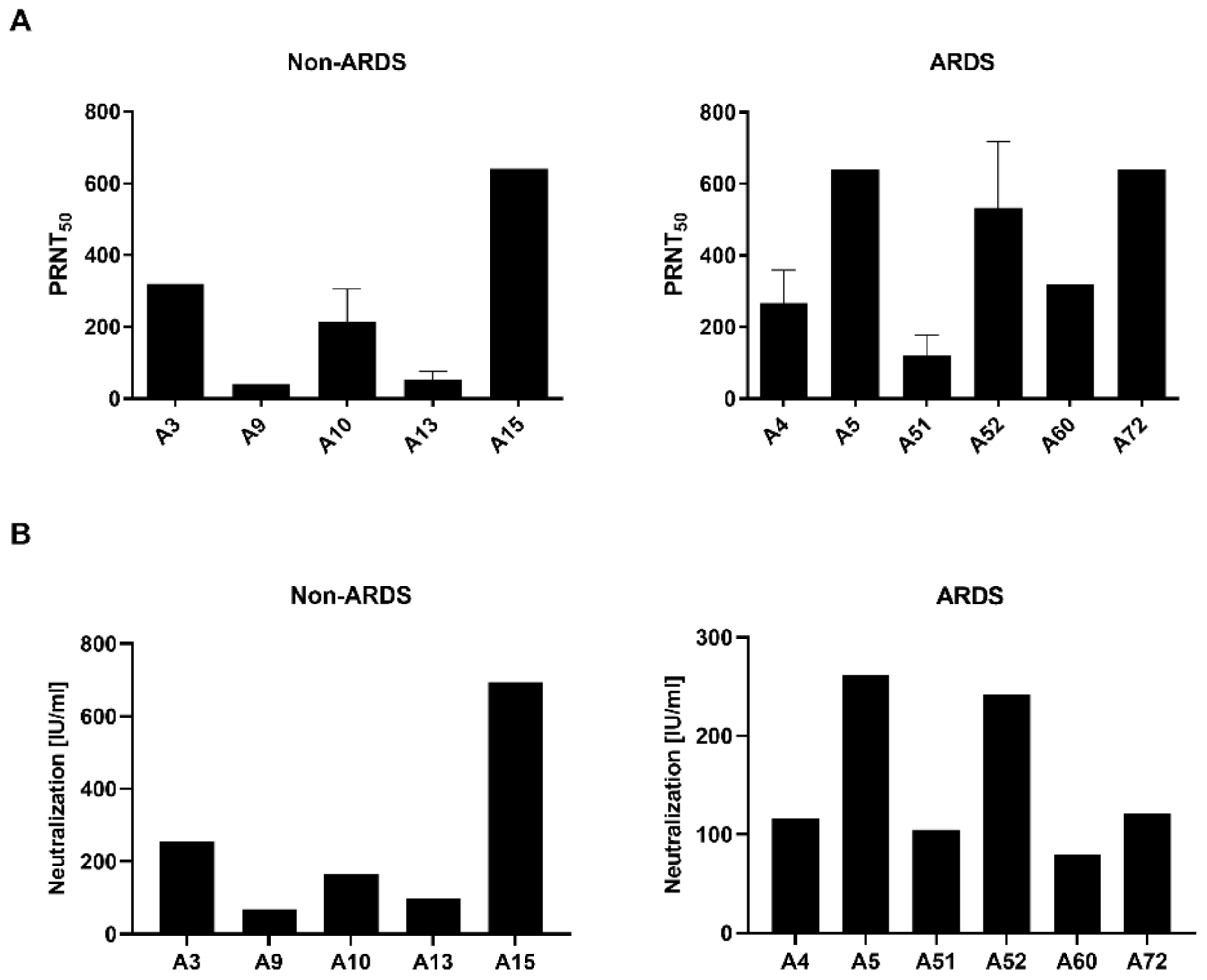
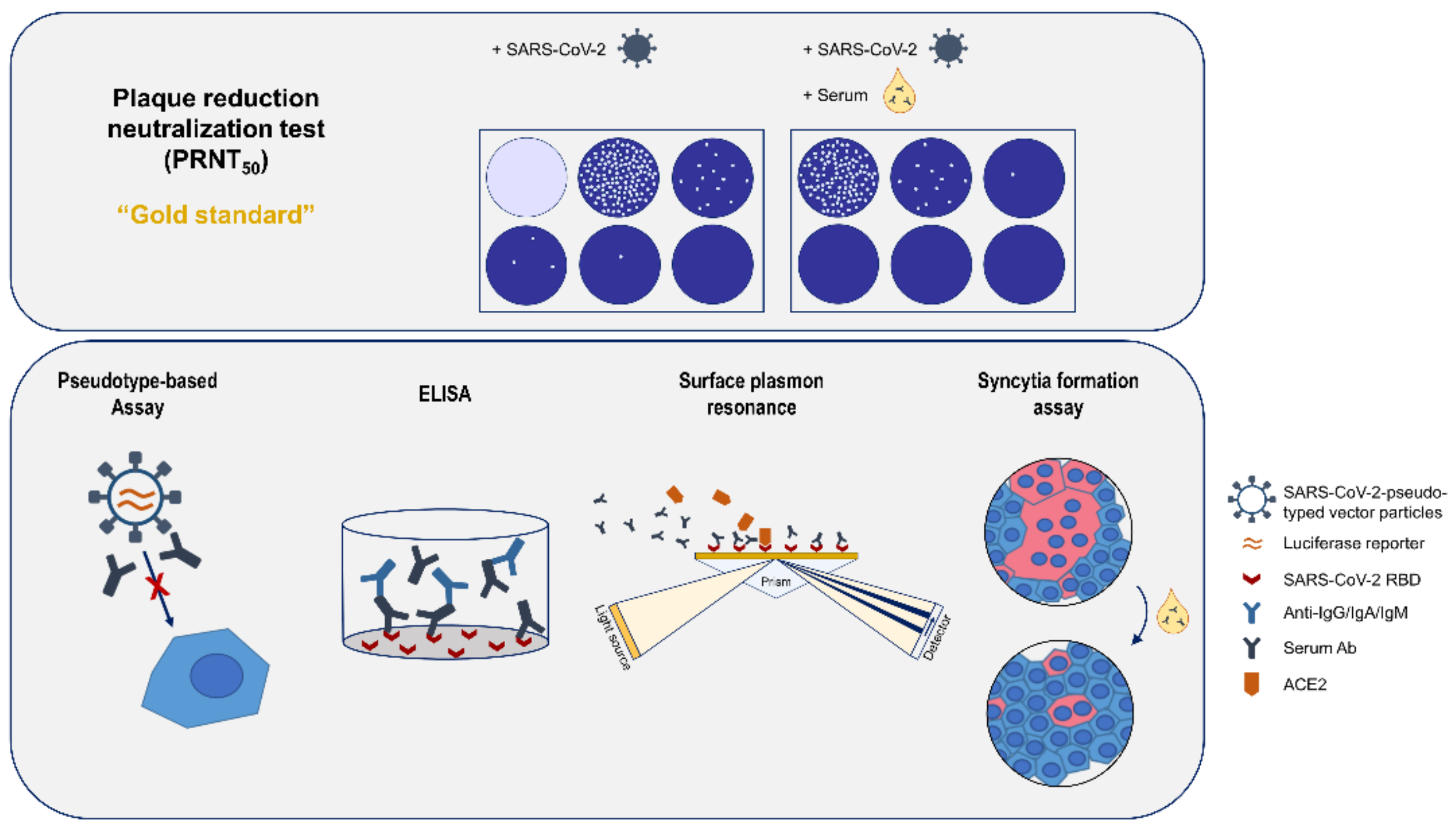
2.3. Plaque Reduction Neutralization Test (PRNT50)
2.4. Pseudotype-Based Neutralization Assay
2.5. Sera
2.6. Ethics
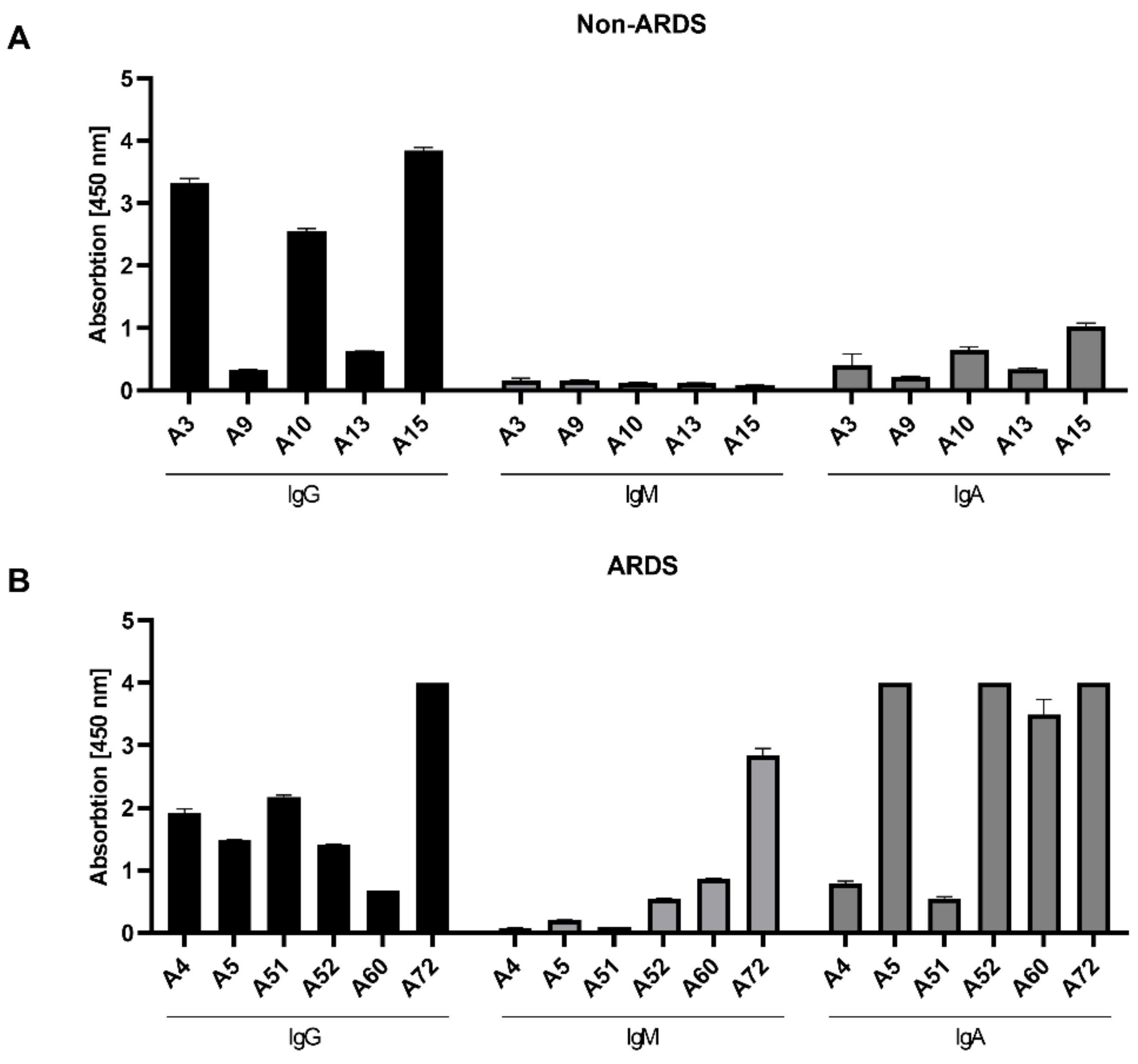
2.7. ELISA
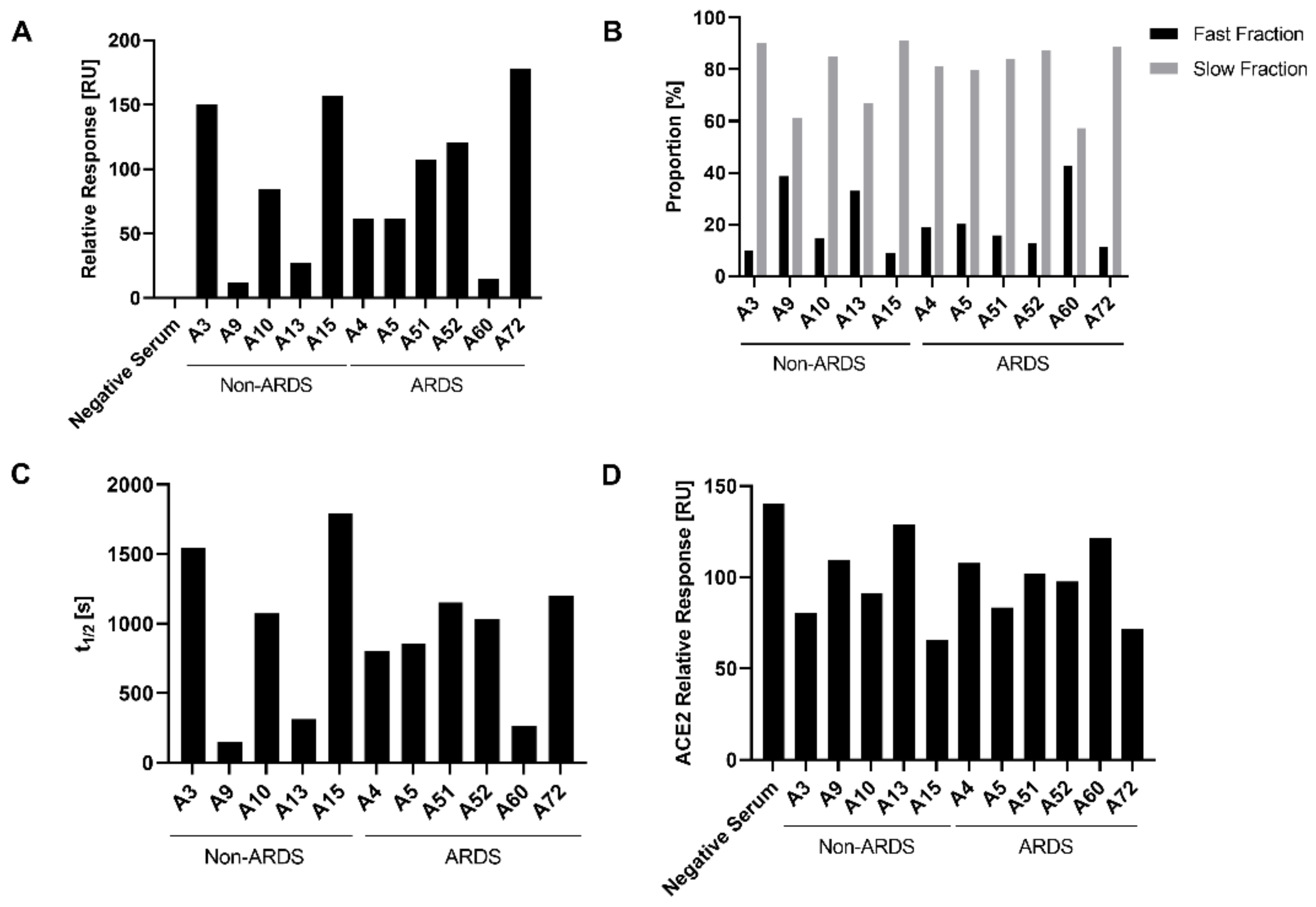
2.8. Surface Plasmon Resonance
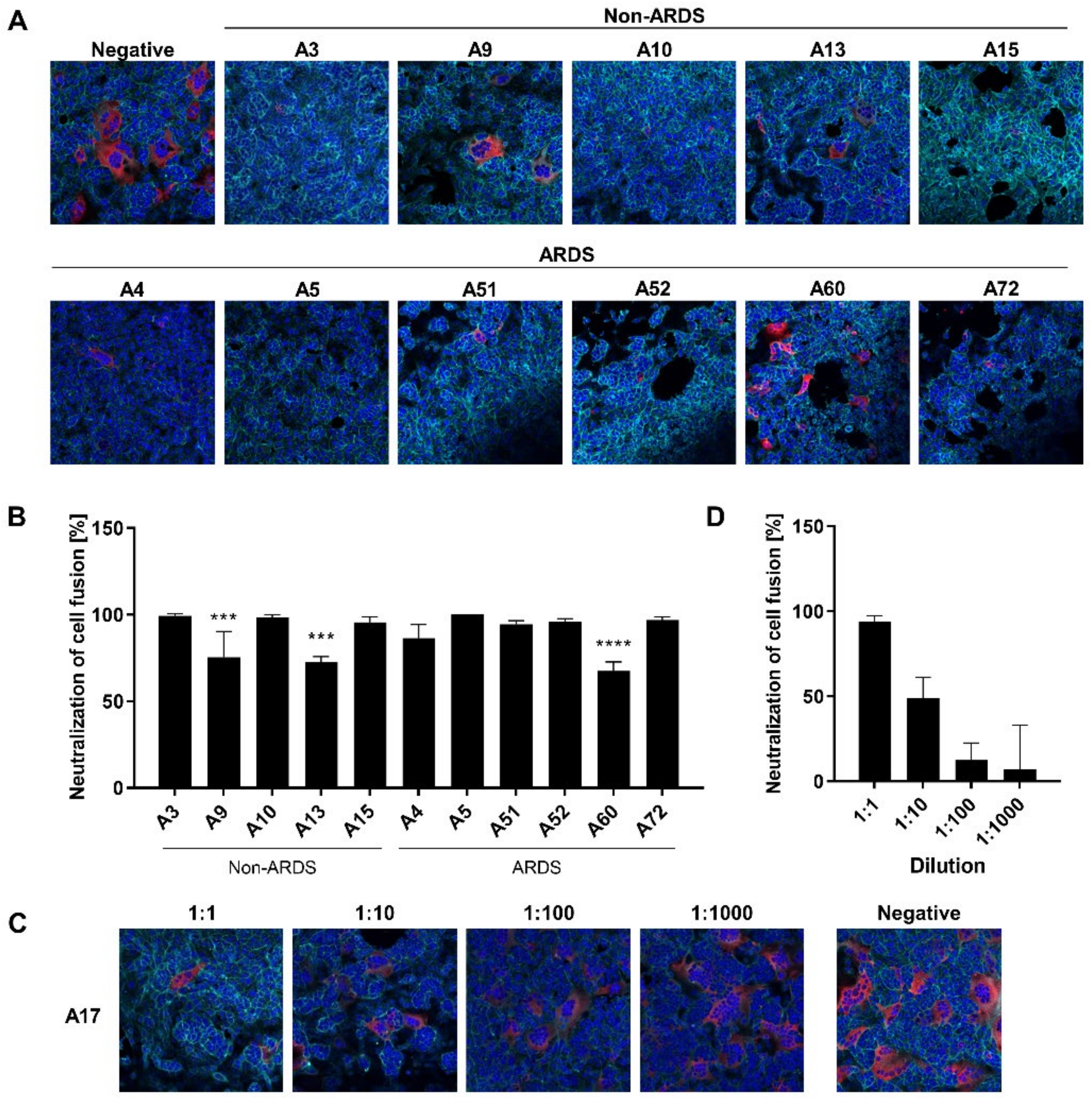
2.9. Cell–Cell Fusion Assay
2.10. Statistical Analysis
2.11. Overview
3. Results
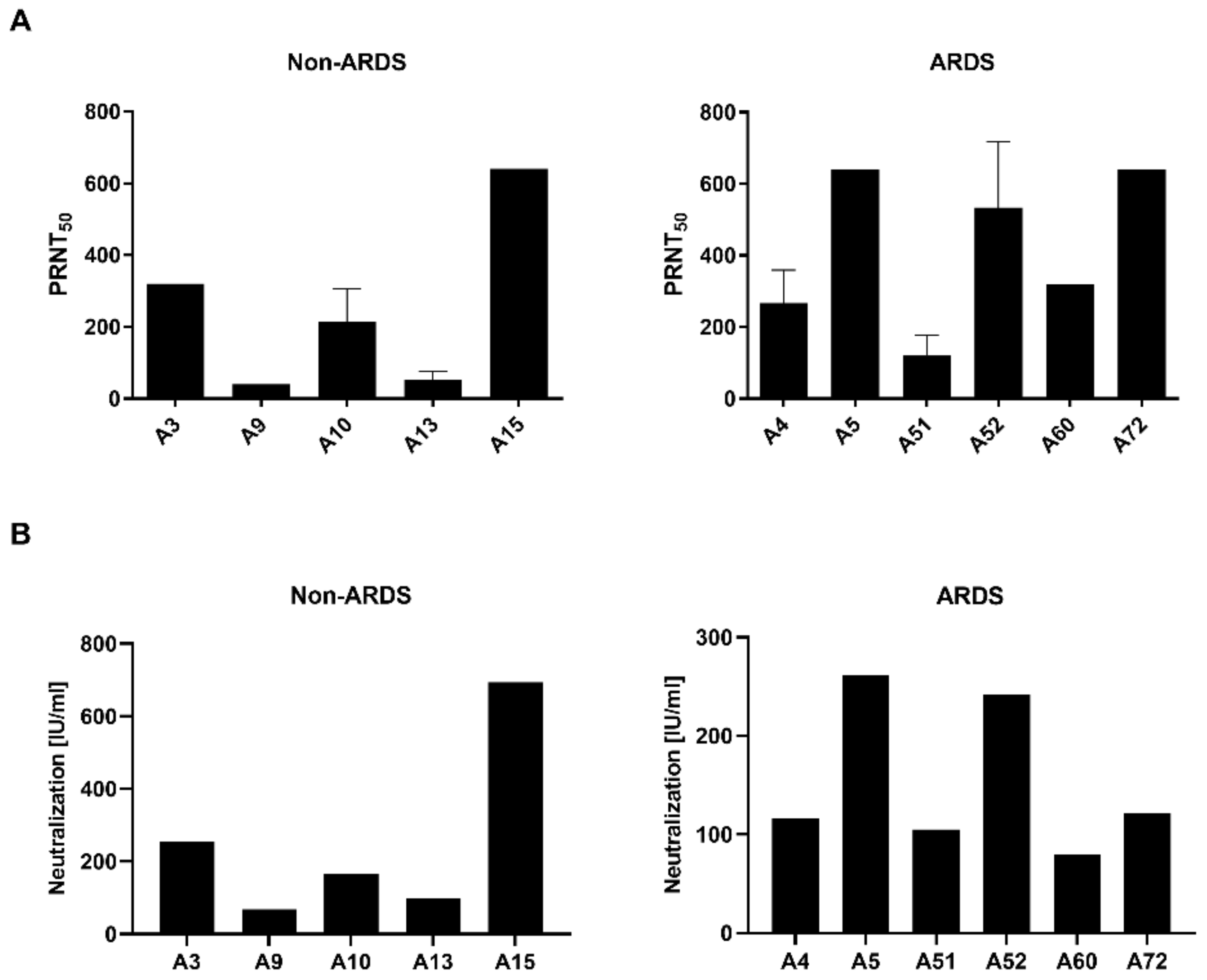
3.1. Quantification of Neutralizating Antibodies by Plaque Reduction Neutralization Test
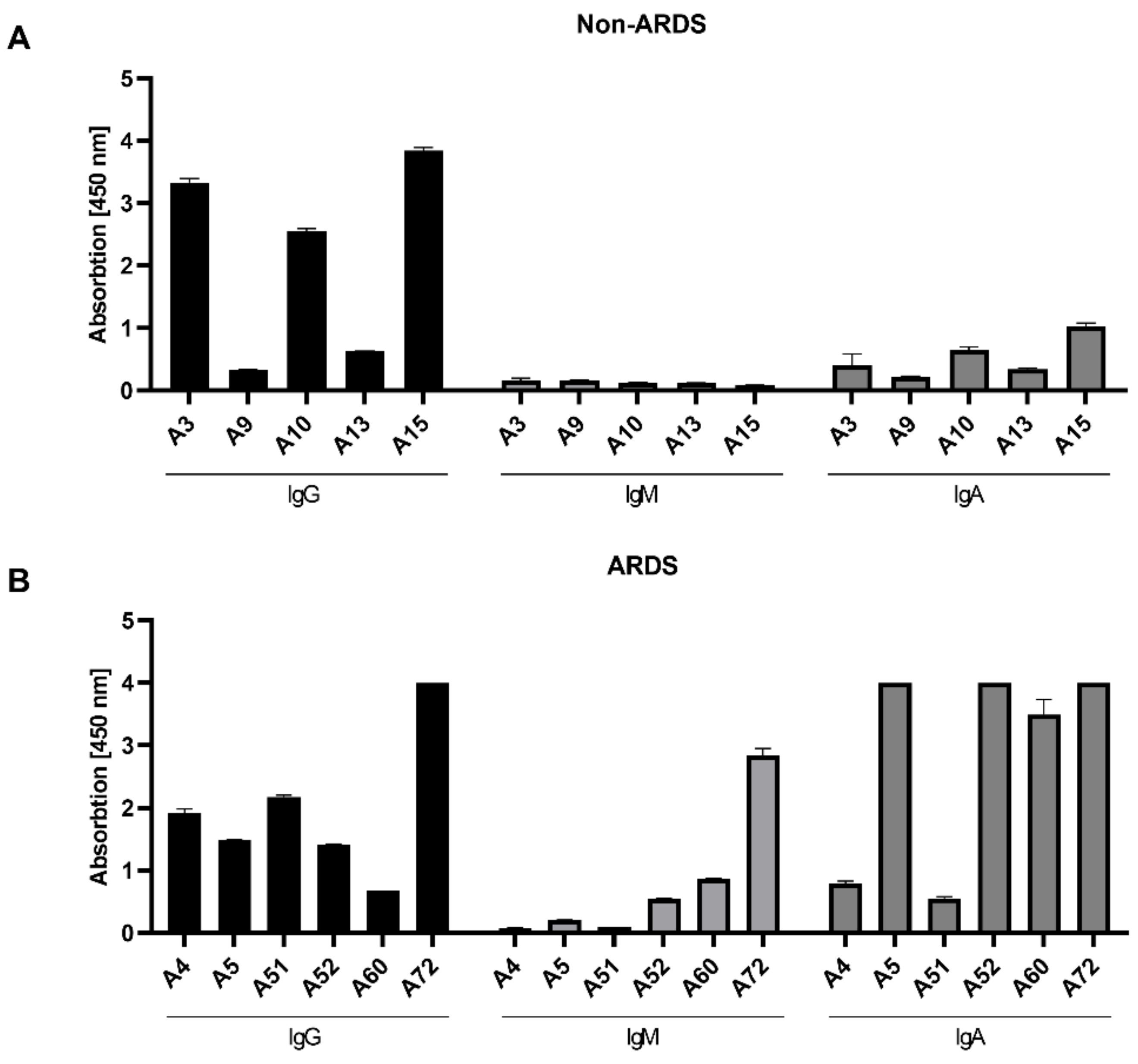
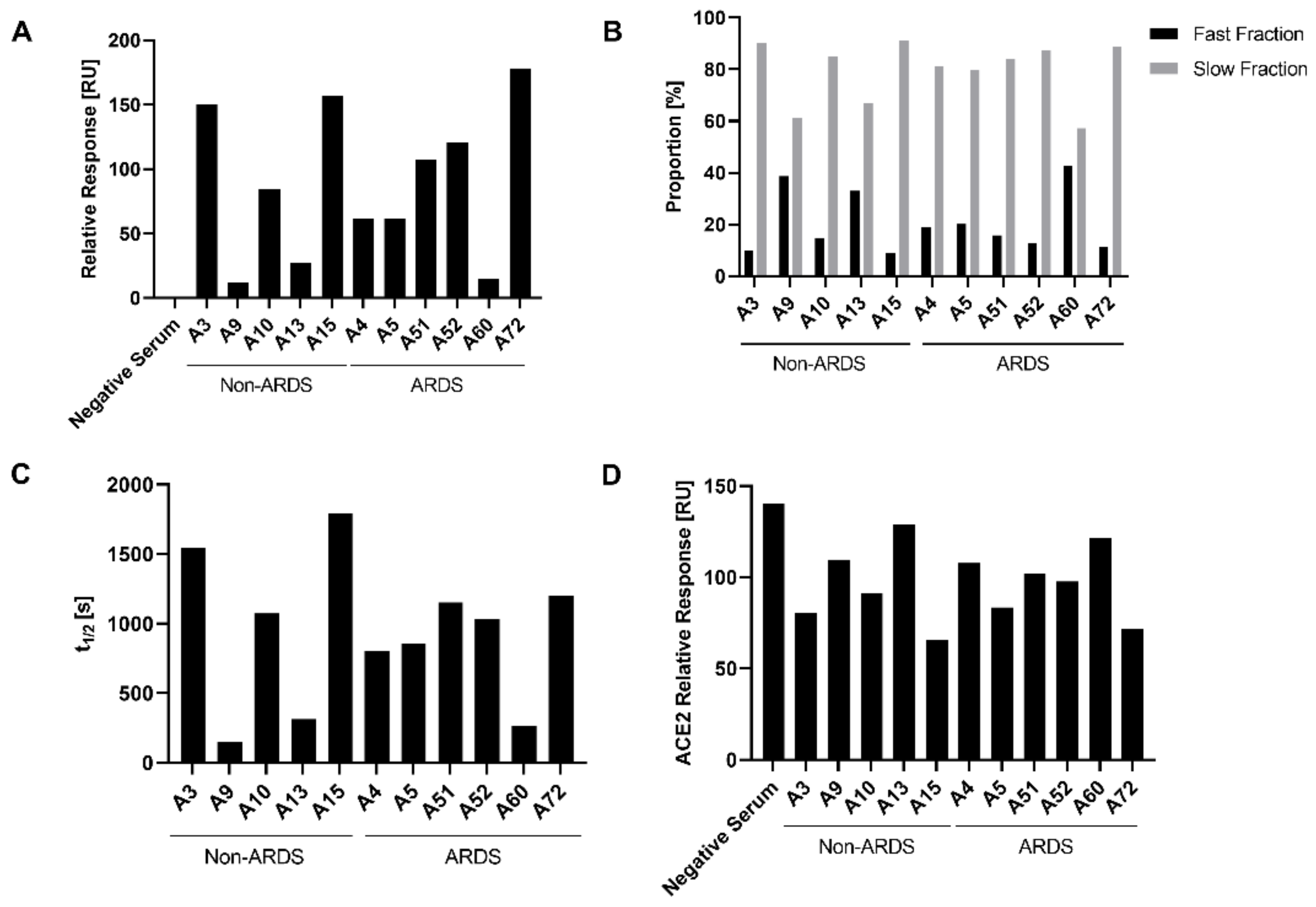
3.2. Class-Specific Quantification of RBD-Specific Antibodies by ELISA
3.3. Analysis of Antibody Affinity to the RBD by Surface Plasmon Resonance
3.4. Characterization of the Impact of Immune Sera on the RBD-ACE-2 Interaction by Surface Plasmon Resonance
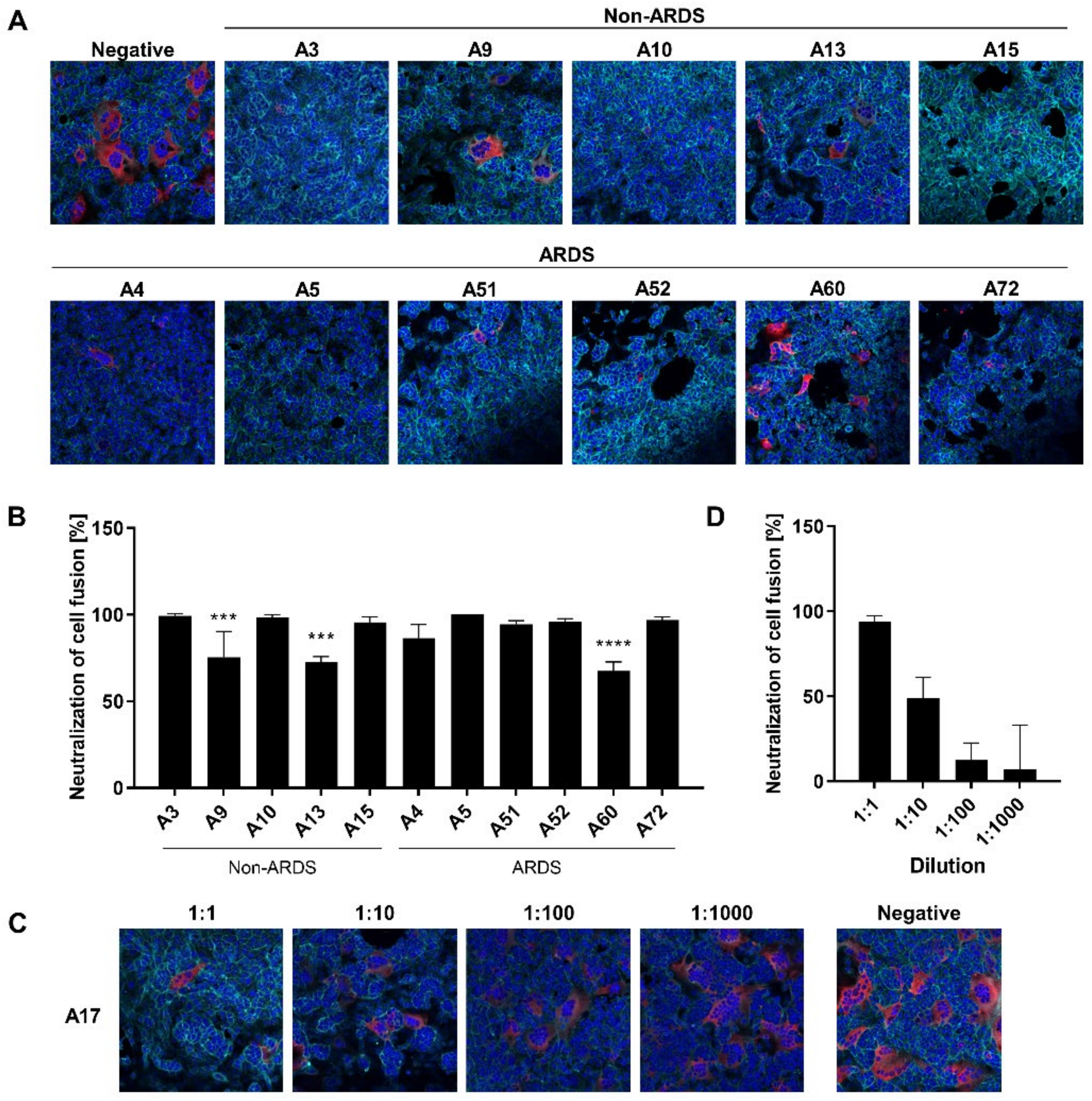
3.5. Inhibition of Syncytium Formation as a Tool to Characterize Spike-Specific Neutralizing Antisera
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. Adv. Virus Res. 2019, 105, 93–116. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, G.; Guo, Y.; Zhou, T.; Gorman, J.; Lee, M.; Rapp, M.; Reddem, E.R.; Yu, J.; Bahna, F.; Bimela, J.; et al. Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite. Cell Host Microbe 2021, 29, 819–833. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; de Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347. [Google Scholar] [CrossRef]
- Schmidt, F.; Weisblum, Y.; Rutkowska, M.; Poston, D.; DaSilva, J.; Zhang, F.; Bednarski, E.; Cho, A.; Schaefer-Babajew, D.J.; Gaebler, C.; et al. High genetic barrier to SARS-CoV-2 polyclonal neutralizing antibody escape. Nature 2021, 600, 512–516. [Google Scholar] [CrossRef]
- Boni, C.; Cavazzini, D.; Bolchi, A.; Rossi, M.; Vecchi, A.; Tiezzi, C.; Barili, V.; Fisicaro, P.; Ferrari, C.; Ottonello, S. Degenerate CD8 Epitopes Mapping to Structurally Constrained Regions of the Spike Protein: A T Cell-Based Way-Out From the SARS-CoV-2 Variants Storm. Front. Immunol. 2021, 12, 730051. [Google Scholar] [CrossRef]
- Galipeau, Y.; Greig, M.; Liu, G.; Driedger, M.; Langlois, M.-A. Humoral Responses and Serological Assays in SARS-CoV-2 Infections. Front. Immunol. 2020, 11, 610688. [Google Scholar] [CrossRef]
- Krammer, F. A correlate of protection for SARS-CoV-2 vaccines is urgently needed. Nat. Med. 2021, 27, 1147–1148. [Google Scholar] [CrossRef]
- Bewley, K.R.; Coombes, N.S.; Gagnon, L.; McInroy, L.; Baker, N.; Shaik, I.; St-Jean, J.R.; St-Amant, N.; Buttigieg, K.R.; Humphries, H.E.; et al. Quantification of SARS-CoV-2 neutralizing antibody by wild-type plaque reduction neutralization, microneutralization and pseudotyped virus neutralization assays. Nat. Protoc. 2021, 16, 3114–3140. [Google Scholar] [CrossRef] [PubMed]
- Sholukh, A.M.; Fiore-Gartland, A.; Ford, E.S.; Hou, Y.; Tse, L.V.; Lempp, F.A.; Kaiser, H.; Saint Germain, R.; Bossard, E.; Kee, J.J.; et al. Evaluation of SARS-CoV-2 neutralization assays for antibody monitoring in natural infection and vaccine trials. medRxiv 2020. [Google Scholar] [CrossRef]
- Rhein, C.; von Scholz, T.; Henss, L.; Kronstein-Wiedemann, R.; Schwarz, T.; Rodionov, R.N.; Corman, V.M.; Tonn, T.; Schnierle, B.S. Comparison of potency assays to assess SARS-CoV-2 neutralizing antibody capacity in COVID-19 convalescent plasma. J. Virol. Methods 2021, 288, 114031. [Google Scholar] [CrossRef]
- Werner, M.; Pervan, P.; Glück, V.; Zeman, F.; Koller, M.; Burkhardt, R.; Glück, T.; Wenzel, J.J.; Schmidt, B.; Gessner, A.; et al. Evaluation of a Broad Panel of SARS-CoV-2 Serological Tests for Diagnostic Use. J. Clin. Med. 2021, 10, 1580. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; van der Werf, S.; et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2020, 39, e106267. [Google Scholar] [CrossRef]
- Theuerkauf, S.A.; Michels, A.; Riechert, V.; Maier, T.J.; Flory, E.; Cichutek, K.; Buchholz, C.J. Quantitative assays reveal cell fusion at minimal levels of SARS-CoV-2 spike protein and fusion from without. iScience 2021, 24, 102170. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.-Y.; Wang, P.; Mok, B.W.-Y.; Zhang, A.J.; Chu, H.; Lee, A.C.-Y.; Deng, S.; Chen, P.; Chan, K.-H.; Song, W.; et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg. Microbes Infect. 2020, 9, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Amanat, F.; Stadlbauer, D.; Strohmeier, S.; Nguyen, T.H.O.; Chromikova, V.; McMahon, M.; Jiang, K.; Arunkumar, G.A.; Jurczyszak, D.; Polanco, J.; et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020, 26, 1033–1036. [Google Scholar] [CrossRef]
- World Health Organization, Initiative for Vaccine Research of the Department of Immunization, Vaccines and Biologicals. Guidelines for Plaque Reduction Neutralization Testing of Human Antibodies to Dengue Viruses. Available online: http://apps.who.int/iris/bitstream/handle/10665/69687/who_ivb_07.07_eng.pdf;jsessionid=A8F694892CAB80CD27AA78C7A0F8540D?sequence=1 (accessed on 31 December 2021).
- Henss, L.; Yue, C.; Kandler, J.; Faddy, H.M.; Simmons, G.; Panning, M.; Lewis-Ximenez, L.L.; Baylis, S.A.; Schnierle, B.S. Establishment of an Alphavirus-Specific Neutralization Assay to Distinguish Infections with Different Members of the Semliki Forest complex. Viruses 2019, 11, 82. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; König, R.; Niedrig, M.; Emmerich, P.; Schnierle, B.S. A neutralization assay for chikungunya virus infections in a multiplex format. J. Virol. Methods 2014, 201, 7–12. [Google Scholar] [CrossRef]
- Henss, L.; Scholz, T.; von Rhein, C.; Wieters, I.; Borgans, F.; Eberhardt, F.J.; Zacharowski, K.; Ciesek, S.; Rohde, G.; Vehreschild, M.; et al. Analysis of Humoral Immune Responses in Patients With Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J. Infect. Dis. 2021, 223, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Cervia, C.; Nilsson, J.; Zurbuchen, Y.; Valaperti, A.; Schreiner, J.; Wolfensberger, A.; Raeber, M.E.; Adamo, S.; Weigang, S.; Emmenegger, M.; et al. Systemic and mucosal antibody responses specific to SARS-CoV-2 during mild versus severe COVID-19. J. Allergy Clin. Immunol. 2021, 147, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo-Campos, P.; Blankenhaus, B.; Mota, C.; Gomes, A.; Serrano, M.; Ariotti, S.; Costa, C.; Nunes-Cabaço, H.; Mendes, A.M.; Gaspar, P.; et al. Seroprevalence of anti-SARS-CoV-2 antibodies in COVID-19 patients and healthy volunteers up to 6 months post disease onset. Eur. J. Immunol. 2020, 50, 2025–2040. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, T.; Weißbrich, B.; Wedekink, F.; Notz, Q.; Herrmann, J.; Krone, M.; Sitter, M.; Schmid, B.; Kredel, M.; Stumpner, J.; et al. Biodistribution and serologic response in SARS-CoV-2 induced ARDS: A cohort study. PLoS ONE 2020, 15, e0242917. [Google Scholar] [CrossRef]
- Tang, J.; Ravichandran, S.; Lee, Y.; Grubbs, G.; Coyle, E.M.; Klenow, L.; Genser, H.; Golding, H.; Khurana, S. Antibody affinity maturation and plasma IgA associate with clinical outcome in hospitalized COVID-19 patients. Nat. Commun. 2021, 12, 1221. [Google Scholar] [CrossRef]
- Yang, G.; Velgos, S.N.; Boddapati, S.P.; Sierks, M.R. Probing Antibody–antigen Interactions. Microbiol. Spectr. 2014, 2, AID-0010-2013. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef]
- Pinto, D.; Sauer, M.M.; Czudnochowski, N.; Low, J.S.; Tortorici, M.A.; Housley, M.P.; Noack, J.; Walls, A.C.; Bowen, J.E.; Guarino, B.; et al. Broad betacoronavirus neutralization by a stem helix-specific human antibody. Science 2021, 373, 1109–1116. [Google Scholar] [CrossRef]
- Sauer, M.M.; Tortorici, M.A.; Park, Y.-J.; Walls, A.C.; Homad, L.; Acton, O.J.; Bowen, J.E.; Wang, C.; Xiong, X.; de van der Schueren, W.; et al. Structural basis for broad coronavirus neutralization. Nat. Struct. Mol. Biol. 2021, 28, 478–486. [Google Scholar] [CrossRef]
- Wohlgemuth, N.; Whitt, K.; Cherry, S.; Kirkpatrick Roubidoux, E.; Lin, C.-Y.; Allison, K.J.; Gowen, A.; Freiden, P.; Allen, E.K.; Gaur, A.H.; et al. An Assessment of Serological Assays for SARS-CoV-2 as Surrogates for Authentic Virus Neutralization. Microbiol. Spectr. 2021, 9, e0105921. [Google Scholar] [CrossRef] [PubMed]
- Padoan, A.; Bonfante, F.; Pagliari, M.; Bortolami, A.; Negrini, D.; Zuin, S.; Bozzato, D.; Cosma, C.; Sciacovelli, L.; Plebani, M. Analytical and clinical performances of five immunoassays for the detection of SARS-CoV-2 antibodies in comparison with neutralization activity. EBioMedicine 2020, 62, 103101. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.N.; Chokkalingam, N.; Reuschel, E.L.; Purwar, M.; Xu, Z.; Gary, E.N.; Kim, K.Y.; Helble, M.; Schultheis, K.; Walters, J.; et al. SARS-CoV-2 Assays To Detect Functional Antibody Responses That Block ACE2 Recognition in Vaccinated Animals and Infected Patients. J. Clin. Microbiol. 2020, 58, e01533-20. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Age | Sex | Group | Symptoms to Hospit. | Days to ICU | ICU Days | Fever Days | Vent Days | Oxygen Days | ECMO Days | Hosp Days |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A3 | 47 | M | non ARDS | 7 | n/a | n/a | 10 | n/a | 25 | n/a | 28 |
| A4 | 69 | W | ARDS | 0 | 3 | 19 | 11 | 14 | 31 | n/a | 39 |
| A5 | 58 | M | ARDS | 5 | 7 | 37 | 40 | 27 | 33 | n/a | 42 |
| A9 | 83 | M | non ARDS | 2 | n/a | n/a | 0 | n/a | 15 | n/a | 28 |
| A10 | 82 | M | non ARDS | 8 | n/a | n/a | 8 | n/a | 29 | n/a | 31 |
| A13 | 80 | M | non ARDS | 6 | n/a | n/a | 7 | n/a | 7 | n/a | 18 |
| A15 | 77 | M | non ARDS | 4 | n/a | n/a | 4 | n/a | 11 | n/a | 12 |
| A51 | 67 | M | ARDS | 0 | 2 | 90 | 32 | 90 | 92 | 23 | 92 |
| A52 | 48 | M | ARDS | 5 | 5 | 87 | 10 | 84 | 87 | 81 | 87 |
| A60 | 77 | W | ARDS | 3 | 3 | 34 | 13 | 26 | 34 | n/a | 56 |
| A72 | 27 | M | ARDS | n/a | n/a | n/a | n/a | n/a | n/a | n/a | 44 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrlein, M.-L.; Hein, S.; Zahn, T.; Mhedhbi, I.; Raupach, J.; Husria, Y.; Benz, N.I.; Eisert, J.; Bender, D.; Haberger, V.; et al. Comparative Investigation of Methods for Analysis of SARS-CoV-2-Spike-Specific Antisera. Viruses 2022, 14, 410. https://doi.org/10.3390/v14020410
Herrlein M-L, Hein S, Zahn T, Mhedhbi I, Raupach J, Husria Y, Benz NI, Eisert J, Bender D, Haberger V, et al. Comparative Investigation of Methods for Analysis of SARS-CoV-2-Spike-Specific Antisera. Viruses. 2022; 14(2):410. https://doi.org/10.3390/v14020410
Chicago/Turabian StyleHerrlein, Marie-Luise, Sascha Hein, Tobias Zahn, Ines Mhedhbi, Jan Raupach, Younes Husria, Nuka Ivalu Benz, Jonathan Eisert, Daniela Bender, Vanessa Haberger, and et al. 2022. "Comparative Investigation of Methods for Analysis of SARS-CoV-2-Spike-Specific Antisera" Viruses 14, no. 2: 410. https://doi.org/10.3390/v14020410
APA StyleHerrlein, M.-L., Hein, S., Zahn, T., Mhedhbi, I., Raupach, J., Husria, Y., Benz, N. I., Eisert, J., Bender, D., Haberger, V., Hastert, F. D., Henss, L., Schnierle, B. S., Stingl, J. C., Dreher, M., & Hildt, E. (2022). Comparative Investigation of Methods for Analysis of SARS-CoV-2-Spike-Specific Antisera. Viruses, 14(2), 410. https://doi.org/10.3390/v14020410