
Full Genome of batCoV/MinFul/2018/SriLanka, a Novel Alpha-Coronavirus Detected in Miniopterus fuliginosus, Sri Lanka

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bat Sampling
2.2. Shotgun NGS
2.3. NGS Data Analysis and Full Genome Assembly
2.4. Sequence Analysis and Phylogenetic Reconstruction
2.5. Real-Time RT-PCR Design
3. Results
3.1. NGS, Full Genome Assembly and Gene Organization
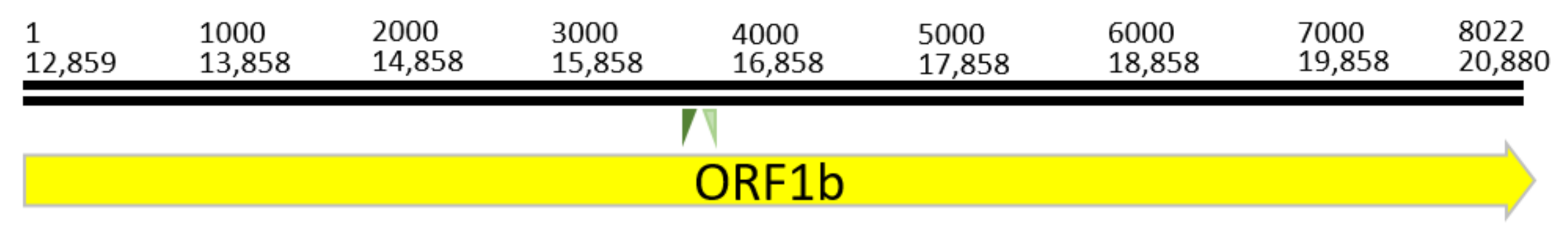
3.1.1. Non-Structural Protein CDS: ORF1a/1b
3.1.2. Structural Protein CDS: Spike, Envelope, Membrane, Nucleotide
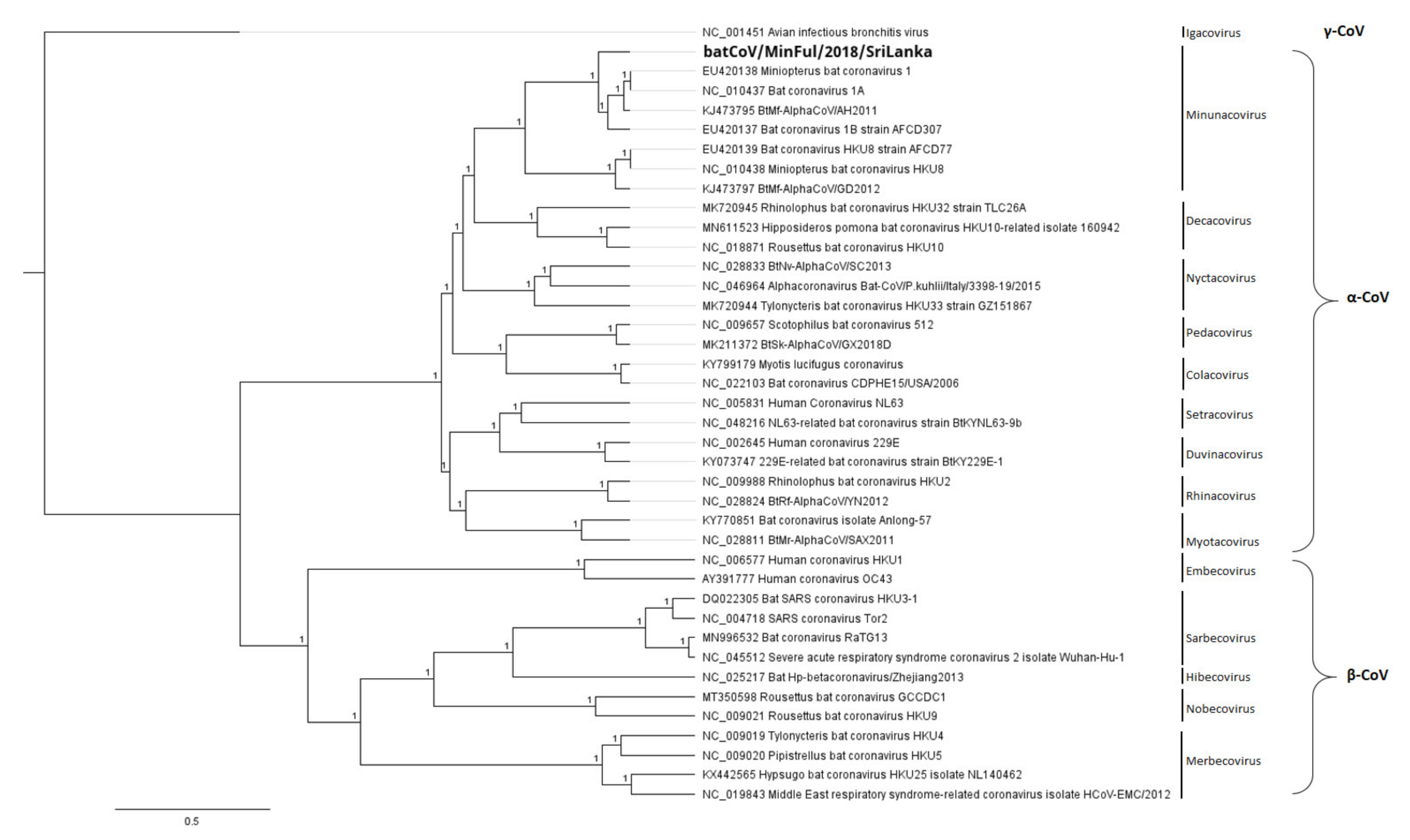
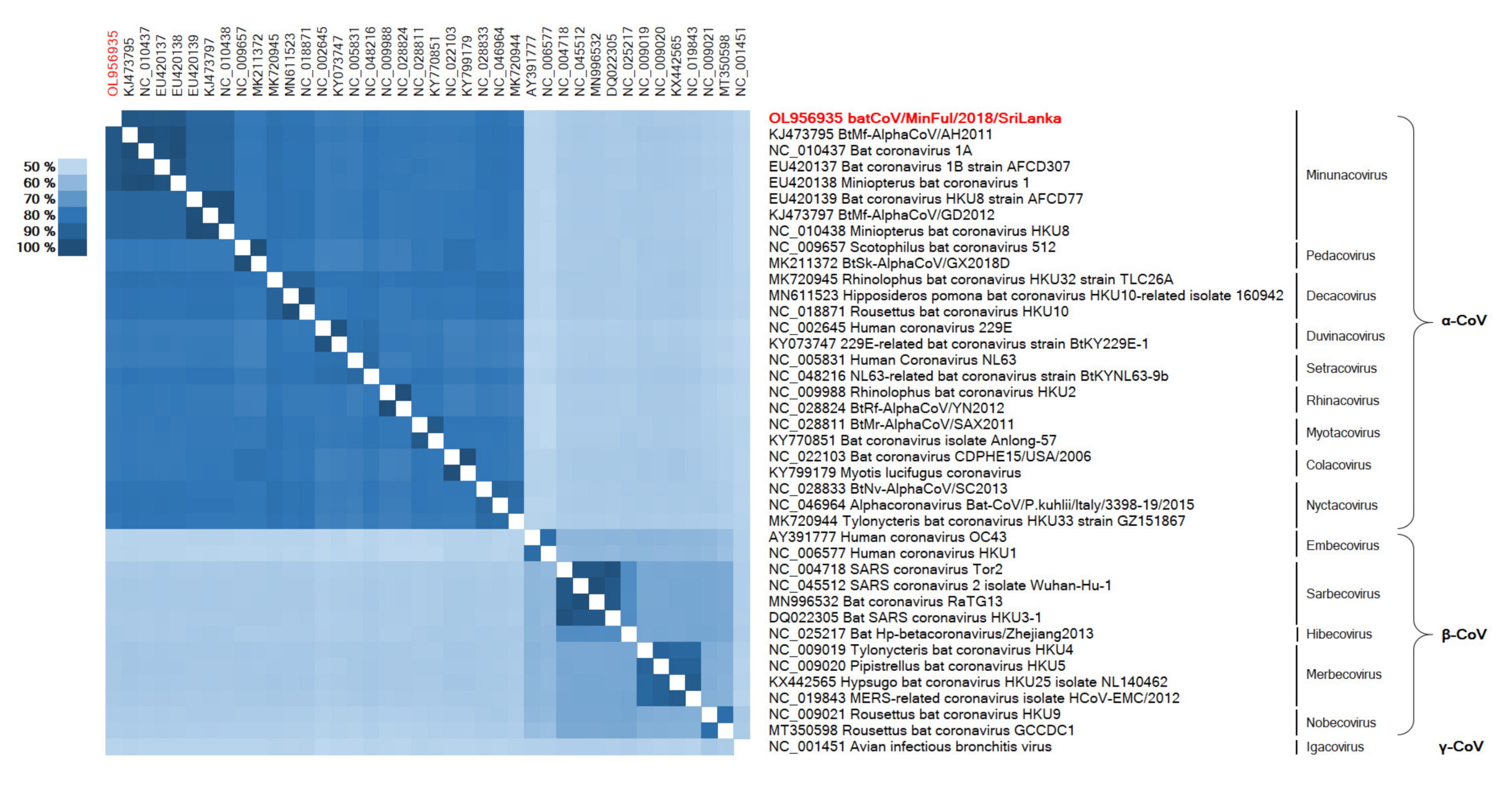
3.2. Phylogeny
3.3. Validation of NGS Results Using Real-Time RT-PCR
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woo, P.C.Y.; Wang, M.; Lau, S.K.P.; Xu, H.; Poon, R.W.S.; Guo, R.; Wong, B.H.L.; Gao, K.; Tsoi, H.W.; Huang, Y.; et al. Comparative Analysis of Twelve Genomes of Three Novel Group 2c and Group 2d Coronaviruses Reveals Unique Group and Subgroup Features. J. Virol. 2007, 81, 1574–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the Largest RNA Virus Genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef] [PubMed]
- González, J.M.; Gomez-Puertas, P.; Cavanagh, D.; Gorbalenya, A.E.; Enjuanes, L. A Comparative Sequence Analysis to Revise the Current Taxonomy of the Family Coronaviridae. Arch. Virol. 2003, 148, 2207–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global Epidemiology of Bat Coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poudel, U.; Subedi, D.; Pantha, S.; Dhakal, S. Animal Coronaviruses and Coronavirus Disease 2019: Lesson for One Health Approach. Open Vet. J. 2020, 10, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, S.M.; Yu, X.H.; Tang, S.L.; Tang, C.K. Coronavirus Disease 2019 (COVID-19): Current Status and Future Perspectives. Int. J. Antimicrob. Agents 2020, 55, 105951. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Yapa, W.B. A Field Guide to the Bats of Sri Lanka, 1st ed.; Dilmah Ceylon Tea Company PLC: Colombo, Sri Lanka, 2017. [Google Scholar]
- Kudagammana, H.D.W.S.; Thevanesam, V.; Chu, D.K.W.; Eriyagama, N.B.; Peiris, J.S.M.; Noordeen, F. Coronaviruses in Guano from Pteropus Medius Bats in Peradeniya, Sri Lanka. Transbound. Emerg. Dis. 2018, 65, 1122–1124. [Google Scholar] [CrossRef] [Green Version]
- Muzeniek, T.; Perera, T.; Siriwardana, S.; Bas, D.; Kaplan, F.; Öruc, M.; Becker-Ziaja, B.; Schwarz, F.; Premawansa, G.; Premawansa, S.; et al. Detection of Alpha-and Betacoronaviruses in Miniopterus Fuliginosus and Rousettus Leschenaultii, Two Species of Sri Lankan Bats. Vaccines 2021, 9, 650. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN Analysis of Metagenomic Data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying Bacterial Genes and Endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved Microbial Gene Identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.L.; Delcher, A.L.; Kasif, S.; White, O. Microbial Gene Identification Using Interpolated Markov Models. Nucleic Acids Res. 1998, 26, 544–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minin, V.N.; Dorman, K.S.; Fang, F.; Suchard, M.A. Dual Multiple Change-Point Model Leads to More Accurate Recombination Detection. Bioinformatics 2005, 21, 3034–3042. [Google Scholar] [CrossRef]
- Sinsheimer, J.S.; Suchard, M.A.; Dorman, K.S.; Fang, F.; Weiss, R.E. Are You My Mother? Bayesian Phylogenetic Inference of Recombination among Putative Parental Strains. Appl. Bioinform. 2003, 2, 131–144. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian Inference of Phylogenetic Trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Baranov, P.V.; Henderson, C.M.; Anderson, C.B.; Gesteland, R.F.; Atkins, J.F.; Howard, M.T. Programmed Ribosomal Frameshifting in Decoding the SARS-CoV Genome. Virology 2005, 332, 498–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namy, O.; Moran, S.J.; Stuart, D.I.; Gilbert, R.J.C.; Brierley, I. A Mechanical Explanation of RNA Pseudoknot Function in Programmed Ribosomal Frameshifting. Nature 2006, 441, 244–247. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Lauber, C.; Siddell, S. Taxonomy of Viruses. Ref. Modul. Biomed. Sci. 2019. [Google Scholar] [CrossRef]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses; Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; et al. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Ge, X.; Wang, L.-F.; Shi, Z. Bat Origin of Human Coronaviruses. Virol. J. 2015, 12, 221. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.F.; Cowled, C. (Eds.) Bats and Viruses: A New Frontier of Emerging Infectious Diseases; Wiley/Blackwell: Hoboken, NJ, USA, 2015; pp. 1–368. [Google Scholar] [CrossRef]
- Kumar, N.; Kaushik, R.; Tennakoon, C.; Uversky, V.N.; Mishra, A.; Sood, R.; Srivastava, P.; Tripathi, M.; Zhang, K.Y.J.; Bhatia, S. Evolutionary Signatures Governing the Codon Usage Bias in Coronaviruses and Their Implications for Viruses Infecting Various Bat Species. Viruses 2021, 13, 1847. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, T.M.; Buchmeier, M.J. Coronavirus Spike Proteins in Viral Entry and Pathogenesis. Virology 2001, 279, 371–374. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhao, S. Furin Cleavage Sites Naturally Occur in Coronaviruses. Stem Cell Res. 2021, 50, 102115. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Sparks, J.S.; Eckerle, L.D.; Sims, A.C.; Denison, M.R. SARS Coronavirus Replicase Proteins in Pathogenesis. Virus Res. 2008, 133, 88–100. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Subgenus | Accession No. | Description |
|---|---|---|---|
| α-CoV | Minunacovirus | KJ473795 | BtMf-AlphaCoV/AH2011 |
| NC_010437 | Bat coronavirus 1A | ||
| EU420137 | Bat coronavirus 1B strain AFCD307 | ||
| EU420138 | Miniopterus bat coronavirus 1 | ||
| EU420139 | Bat coronavirus HKU8 strain AFCD77 | ||
| KJ473797 | BtMf-AlphaCoV/GD2012 | ||
| NC_010438 | Miniopterus bat coronavirus HKU8 | ||
| Pedacovirus | NC_009657 | Scotophilus bat coronavirus 512 | |
| MK211372 | BtSk-AlphaCoV/GX2018D | ||
| Decacovirus | MK720945 | Rhinolophus bat coronavirus HKU32 strain TLC26A | |
| MN611523 | Hipposideros pomona bat coronavirus HKU10-related isolate 160942 | ||
| NC_018871 | Rousettus bat coronavirus HKU10 | ||
| Duvinacovirus | NC_002645 | Human coronavirus 229E | |
| KY073747 | 229E-related bat coronavirus strain BtKY229E-1 | ||
| Setracovirus | NC_005831 | Human coronavirus NL63 | |
| NC_048216 | NL63-related bat coronavirus strain BtKYNL63-9b | ||
| Rhinacovirus | NC_009988 | Rhinolophus bat coronavirus HKU2 | |
| NC_028824 | BtRf-AlphaCoV/YN2012 | ||
| Myotacovirus | NC_028811 | BtMr-AlphaCoV/SAX2011 | |
| KY770851 | Bat coronavirus isolate Anlong-57 | ||
| Colacovirus | NC_022103 | Bat coronavirus CDPHE15/USA/2006 | |
| KY799179 | Myotis lucifugus coronavirus | ||
| Nyctacovirus | NC_028833 | BtNv-AlphaCoV/SC2013 | |
| NC_046964 | Alphacoronavirus bat-CoV/P.kuhlii/Italy/3398-19/2015 | ||
| MK720944 | Tylonycteris bat coronavirus HKU33 strain GZ151867 | ||
| β-CoV | Embecovirus | AY391777 | Human coronavirus OC43 |
| NC_006577 | Human coronavirus HKU1 | ||
| Sarbecovirus | NC_004718 | SARS coronavirus Tor2 | |
| NC_045512 | SARS coronavirus 2 isolate Wuhan-Hu-1 | ||
| MN996532 | Bat coronavirus RaTG13 | ||
| DQ022305 | Bat SARS coronavirus HKU3-1 | ||
| Hibecovirus | NC_025217 | Bat Hp-betacoronavirus/Zhejiang2013 | |
| Merbecovirus | NC_009019 | Tylonycteris bat coronavirus HKU4 | |
| NC_009020 | Pipistrellus bat coronavirus HKU5 | ||
| KX442565 | Hypsugo bat coronavirus HKU25 isolate NL140462 | ||
| NC_019843 | MERS-related coronavirus isolate HCoV-EMC/2012 | ||
| Nobecovirus | NC_009021 | Rousettus bat coronavirus HKU9 | |
| MT350598 | Rousettus bat coronavirus GCCDC1 | ||
| γ-CoV | Igacovirus | NC_001451 | Avian infectious bronchitis virus |
| CDS | Start–End (Nucleotide Position) | No. of Nucleotides | No. of Amino Acids | |
|---|---|---|---|---|
| batCoV/ MinFul/2018/ SriLanka | ORF1a | 113–12,859 | 12,747 | 4249 |
| ORF1b | 12,859–20,880 | 8022 | 2674 | |
| Spike | 20,882–25,009 | 4128 | 1376 | |
| Envelope | 25,662–25,886 | 225 | 75 | |
| Membrane | 25,893–26,651 | 759 | 253 | |
| Nucleocapsid | 26,672–27,841 | 1170 | 390 | |
| KJ473795 | ORF1a | 273–13,046 | 12,774 | 4258 |
| ORF1b | 13,046–21,067 | 8022 | 2674 | |
| Spike | 21,069–25,196 | 4128 | 1376 | |
| Envelope | 25,849–26,073 | 225 | 75 | |
| Membrane | 26,080–26,841 | 762 | 251 | |
| Nucleocapsid | 26,862–28,031 | 1170 | 390 |
| Pairwise Nucleotide Identity (%) | Pairwise Amino Acid Identity (%) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Full Genome | ORF1a | ORF1b | S | E | M | N | ORF1a | ORF1b | S | E | M | N | |
| Alphacoronaviruses | |||||||||||||
| Miniopterus bat coronavirus 1 | 84.9 | 83.0 | 89.2 | 83.1 | 92.4 | 88.1 | 88.5 | 86.1 | 94.9 | 87.1 | 91.9 | 88.7 | 89.5 |
| BtMf-AlphaCoV/AH2011 | 85.1 | 83.2 | 89.7 | 83.2 | 92.9 | 86.5 | 89.2 | 86.6 | 95.5 | 87.7 | 91.9 | 87.5 | 98.7 |
| BtMf-AlphaCoV/GD2012 | 66.8 | 65.5 | 77.7 | 62.5 | 70.7 | 73.9 | 58.0 | 63.3 | 87.4 | 59.1 | 67.6 | 74.5 | 57.2 |
| Human coronavirus 229E | 57.0 | 54.9 | 71.7 | 46.7 | 57.6 | 54.3 | 43.3 | 47.7 | 77.5 | 45.0 | 44.7 | 56.2 | 37.8 |
| Human coronavirus NL63 | 59.4 | 55.7 | 73.3 | 52.8 | 61.5 | 56.0 | 49.3 | 47.7 | 77.6 | 43.5 | 51.3 | 60.6 | 45.5 |
| Betacoronaviruses | |||||||||||||
| Bat SARS coronavirus HKU3-1 | 39.4 | 34.3 | 58.0 | 32.6 | 34.6 | 39.3 | 32.2 | 19.7 | 56.1 | 18.1 | 21.3 | 30.5 | 22.4 |
| Rousettus bat coronavirus HKU9 | 39.1 | 33.2 | 57.9 | 34.0 | 41.5 | 43.6 | 30.6 | 19.7 | 55.8 | 18.3 | 15.6 | 31.6 | 18.5 |
| Pipistrellus bat coronavirus HKU5 | 38.0 | 32.4 | 57.1 | 34.2 | 37.4 | 38.4 | 30.8 | 20.5 | 56.2 | 19.0 | 16.3 | 32.6 | 24.1 |
| Human coronavirus HKU1 | 42.8 | 39.9 | 58.5 | 38.6 | 42.8 | 42.6 | 33.5 | 20.3 | 53.3 | 18.2 | 17.7 | 34.6 | 23.7 |
| SARS-CoV-2 Wuhan-Hu-1 | 40.2 | 34.7 | 59.6 | 34.4 | 35.5 | 39.6 | 31.9 | 20.2 | 56.5 | 18.3 | 20.0 | 30.8 | 21.7 |
| Human coronavirus OC43 | 40.3 | 39.0 | 57.3 | 37.7 | 40.3 | 41.1 | 32.0 | 20.3 | 53.2 | 18.1 | 17.9 | 32.5 | 21.0 |
| Pool | Positive Samples/Total | Positive Sample | Sampling Date | Sex | Copies per Reaction (25 µL) |
|---|---|---|---|---|---|
| RS 2.2 | 6/8 | RS 85 | 7 July 2018 | M | (1) |
| RS 91 | 7 July 2018 | F | (14) | ||
| RS 94 | 7 July 2018 | M | (12) | ||
| RS 95 | 7 July 2018 | F | 1313 | ||
| RS 96 | 7 July 2018 | F | 60 | ||
| RS 98 | 7 July 2018 | F | 468 | ||
| RS 2.3 | 4/8 | RS 106 | 7 July 2018 | F | 106 |
| RS 108 | 7 July 2018 | F | 371 | ||
| RS 109 | 7 July 2018 | M | 854 | ||
| RS 110 | 7 July 2018 | F | 1737 | ||
| RS 2.4 | 4/7 | RS 114 | 7 July 2018 | F | 70 |
| RS 117 | 7 July 2018 | F | 46 | ||
| RS 118 | 7 July 2018 | F | (14) | ||
| RS 119 | 7 July 2018 | F | 650 | ||
| RS 2.5 | 3/7 | RS 124 | 7 July 2018 | F | 105 |
| RS 126 | 7 July 2018 | F | 1391 | ||
| RS 133 | 7 July 2018 | M | 242 | ||
| RS 2.6 | 3/6 | RS 135 | 7 July 2018 | F | 339 |
| RS 137 | 7 July 2018 | F | 2124 | ||
| RS 138 | 7 July 2018 | F | 106 | ||
| RS 2.7 | 2/7 | RS 146 | 7 July 2018 | F | 44 |
| RS 147 | 7 July 2018 | F | 93 | ||
| RS 2.8 | 5/9 | RS 154 | 8 July 2018 | F | 2611 |
| RS 158 | 8 July 2018 | F | 71 | ||
| RS 159 | 8 July 2018 | F | 1196 | ||
| RS 162 | 8 July 2018 | F | 866 | ||
| RS 164 | 8 July 2018 | F | 33 | ||
| RS 2.9 | 7/8 | RS 168 | 8 July 2018 | F | 296 |
| RS 169 | 8 July 2018 | F | 76 | ||
| RS 170 | 8 July 2018 | F | 557 | ||
| RS 171 | 8 July 2018 | M | 196 | ||
| RS 172 | 8 July 2018 | F | 104 | ||
| RS 175 | 8 July 2018 | F | 464 | ||
| RS 176 | 8 July 2018 | F | 753 | ||
| RS 2.10 | 2/5 | RS 178 | 8 July 2018 | F | 600 |
| RS 187 | 8 July 2018 | F | (5) |
| Genus | Miniopterus | Rousettus | Hipposideros | Rhinolophus | ||||
|---|---|---|---|---|---|---|---|---|
| Rectal Swabs | Feces | Rectal Swabs | Feces | Rectal Swabs | Feces | Rectal Swabs | Feces | |
| March 2018 | ||||||||
| Pools | 2 | 0 | 1 | 1 | 1 | 0 | 12 | 1 |
| Positive samples | 3/3 | 0/0 | 0/9 | 0/2 | 0/3 | 0/0 | 0/60 | 0/8 |
| July 2018 | ||||||||
| Pools | 17 | 8 | 1 | 0 | 0 | 0 | 0 | 0 |
| Positive samples | 59/116 | 38/77 | 0/11 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| January 2019 | ||||||||
| Pools | 1 | 3 | 2 | 1 | 3 | 2 | 2 | 2 |
| Positive samples | 4/11 | 23/27 | 0/16 | 0/3 | 0/16 | 0/7 | 0/16 | 0/17 |
| Total positive | 66/130 (50%) | 61/104 (58%) | 0/36 | 0/5 | 0/19 | 0/7 | 0/76 | 0/25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muzeniek, T.; Perera, T.; Siriwardana, S.; Bas, D.; Kaplan, F.; Öruc, M.; Becker-Ziaja, B.; Perera, I.; Weerasena, J.; Handunnetti, S.; et al. Full Genome of batCoV/MinFul/2018/SriLanka, a Novel Alpha-Coronavirus Detected in Miniopterus fuliginosus, Sri Lanka. Viruses 2022, 14, 337. https://doi.org/10.3390/v14020337
Muzeniek T, Perera T, Siriwardana S, Bas D, Kaplan F, Öruc M, Becker-Ziaja B, Perera I, Weerasena J, Handunnetti S, et al. Full Genome of batCoV/MinFul/2018/SriLanka, a Novel Alpha-Coronavirus Detected in Miniopterus fuliginosus, Sri Lanka. Viruses. 2022; 14(2):337. https://doi.org/10.3390/v14020337
Chicago/Turabian StyleMuzeniek, Therese, Thejanee Perera, Sahan Siriwardana, Dilara Bas, Fatimanur Kaplan, Mizgin Öruc, Beate Becker-Ziaja, Inoka Perera, Jagathpriya Weerasena, Shiroma Handunnetti, and et al. 2022. "Full Genome of batCoV/MinFul/2018/SriLanka, a Novel Alpha-Coronavirus Detected in Miniopterus fuliginosus, Sri Lanka" Viruses 14, no. 2: 337. https://doi.org/10.3390/v14020337
APA StyleMuzeniek, T., Perera, T., Siriwardana, S., Bas, D., Kaplan, F., Öruc, M., Becker-Ziaja, B., Perera, I., Weerasena, J., Handunnetti, S., Schwarz, F., Premawansa, G., Premawansa, S., Yapa, W., Nitsche, A., & Kohl, C. (2022). Full Genome of batCoV/MinFul/2018/SriLanka, a Novel Alpha-Coronavirus Detected in Miniopterus fuliginosus, Sri Lanka. Viruses, 14(2), 337. https://doi.org/10.3390/v14020337