
Viral Proteins with PxxP and PY Motifs May Play a Role in Multiple Sclerosis




Abstract
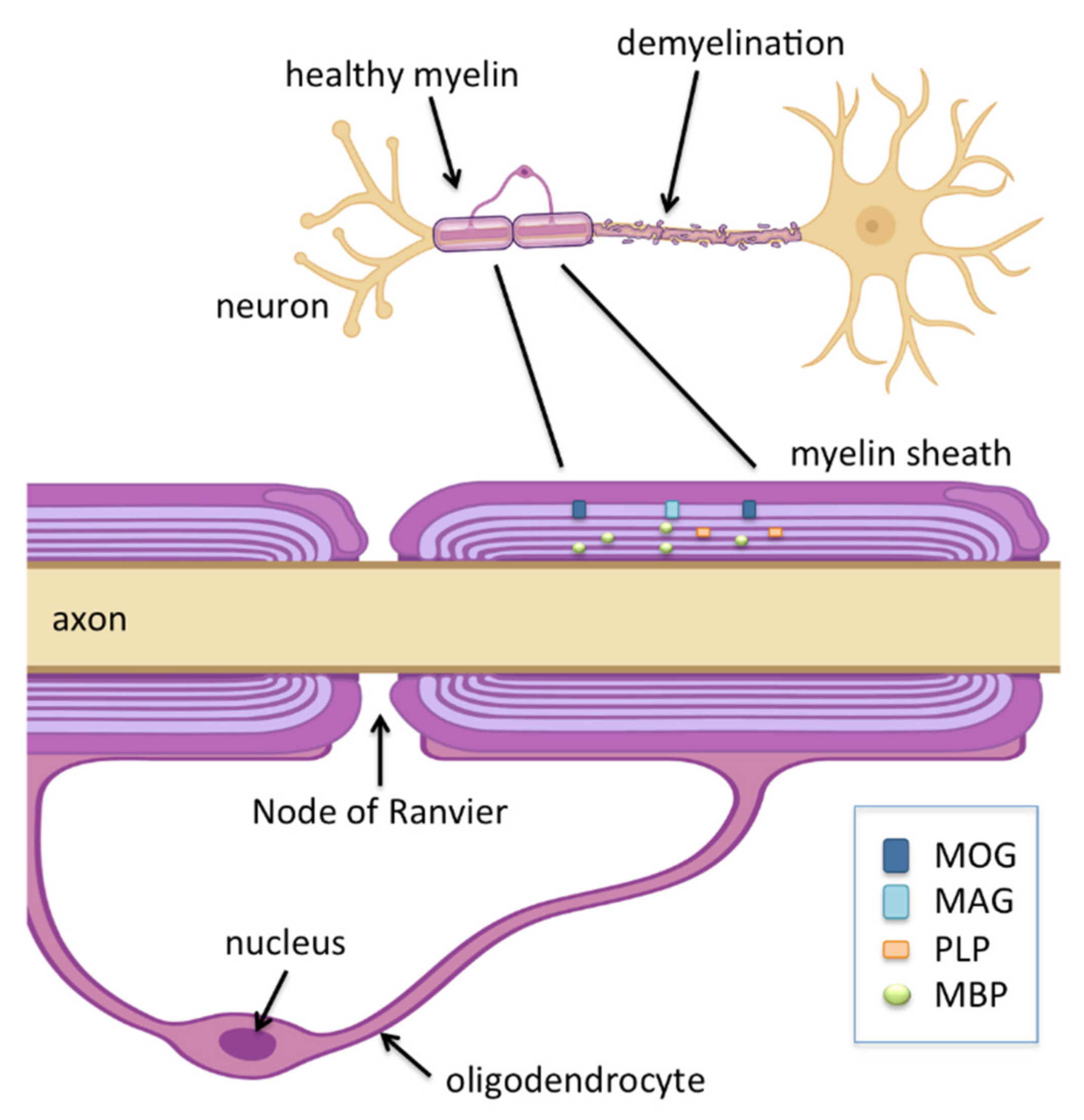
1. Multiple Sclerosis
2. Viral Risk Factors
2.1. Epstein–Barr Virus (EBV)
2.2. Human Herpes Virus Type 6 (HHV-6)
2.3. Cytomegalovirus (CMV)
2.4. Other Viruses
3. Proline-Rich Proteins and Their Ligands

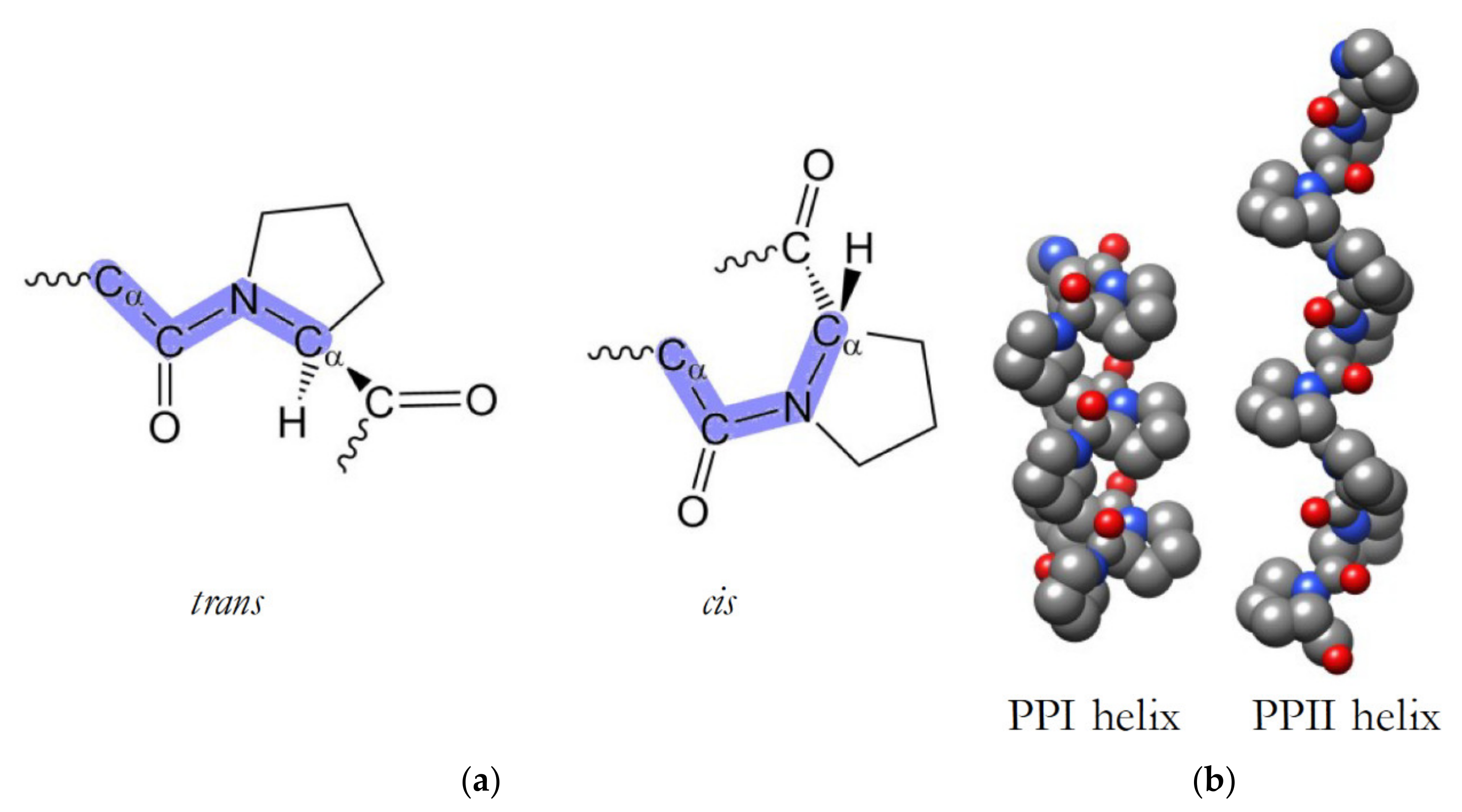
3.1. PxxP Motifs Bind to Src Homology 3 (SH3) Domains
3.2. PPxY (PY) Motifs Bind to WW Domains
4. SH3 and WW Domains Implicated in MS
4.1. MBP/Fyn-SH3 Interactions
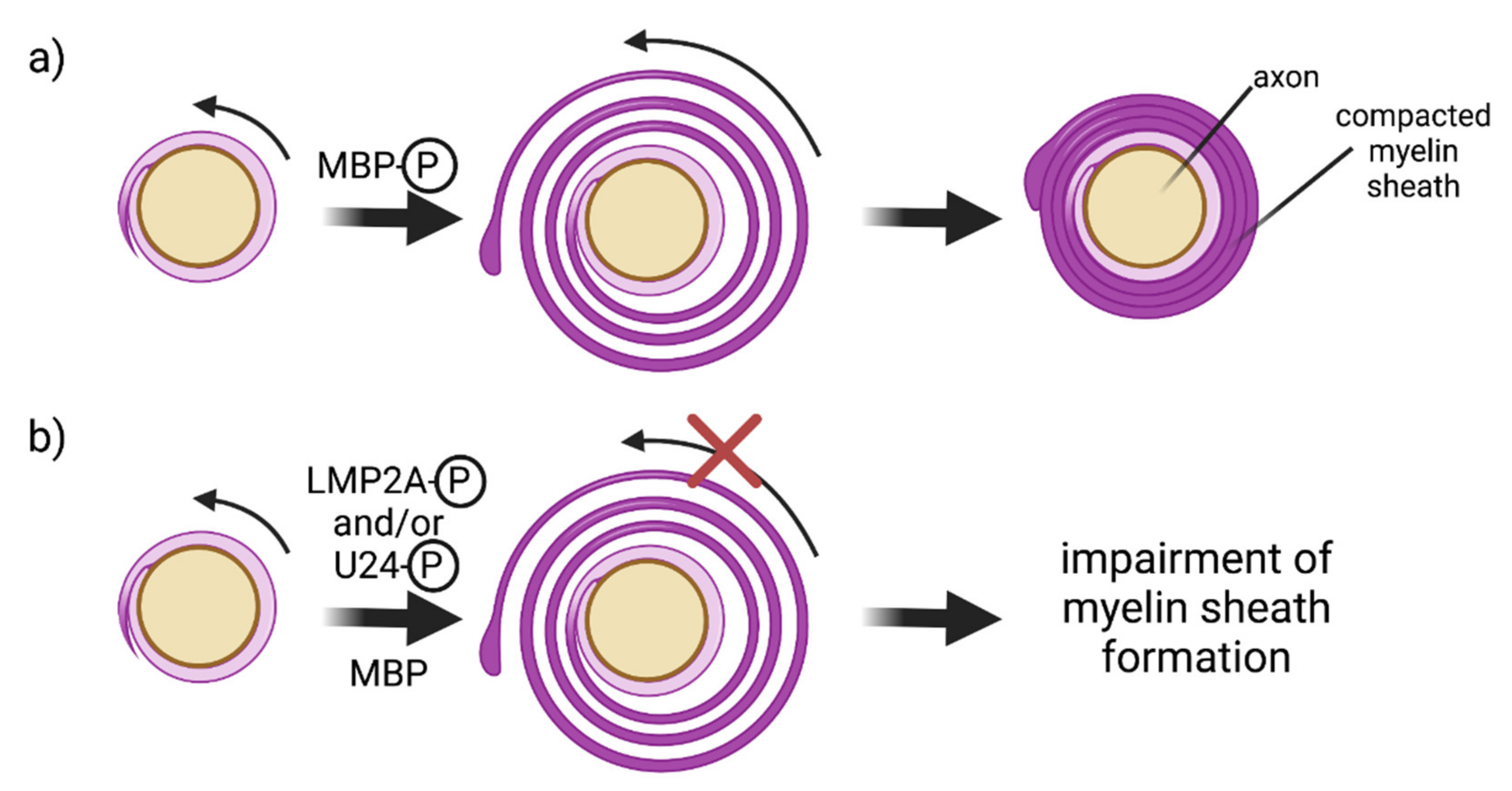
4.2. E3 Ubiquitin Ligases and Oligodendrocytes
5. Phosphorylation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Donati, D.; Jacobson, S. Viruses and multiple sclerosis. In Polymicrobial Diseases; Brogden, K.A., Guthmiller, J.M., Eds.; ASM Press: Washington, DC, USA, 2002. [Google Scholar]
- Lublin, F.D.; Reingold, S.C. Defining the clinical course of multiple sclerosis: Results of an international survey. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.J.; Abu-Rub, M.; Miller, R.H. B Cells in Neuroinflammation: New Perspectives and Mechanistic Insights. Cells 2021, 10, 1605. [Google Scholar] [CrossRef]
- Moazami, F.; Lefevre-Utile, A.; Papaloukas, C.; Soumelis, V. Machine Learning Approaches in Study of Multiple Sclerosis Disease Through Magnetic Resonance Images. Front. Immunol. 2021, 12, 3205. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Coetzee, T.; Cohen, J.A.; Marrie, R.A.; Thompson, A.J. The 2013 clinical course descriptors for multiple sclerosis: A clarification. Neurology 2020, 94, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front. Immunol. 2019, 10, 3116. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Pender, M.P.; Khanna, R.; Steinman, L.; Hartung, H.P.; Maniar, T.; Croze, E.; Aftab, B.T.; Giovannoni, G.; Joshi, M.J. Epstein–Barr Virus in Multiple Sclerosis: Theory and Emerging Immunotherapies. Trends Mol. Med. 2020, 26, 296–310. [Google Scholar] [CrossRef]
- Tarlinton, R.E.; Martynova, E.; Rizvanov, A.A.; Khaiboullina, S.; Verma, S. Role of viruses in the pathogenesis of multiple sclerosis. Viruses 2020, 12, 643. [Google Scholar] [CrossRef] [PubMed]
- Brummer, T.; Ruck, T.; Meuth, S.G.; Zipp, F.; Bittner, S. Treatment approaches to patients with multiple sclerosis and coexisting autoimmune disorders. Ther. Adv. Vaccines 2021, 14, 17562864211035542. [Google Scholar] [CrossRef] [PubMed]
- Jalkh, G.; Abi Nahed, R.; Macaron, G.; Rensel, M. Safety of newer disease modifying therapies in multiple sclerosis. Vaccines 2021, 9, 12. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Multiple sclerosis review. Pharm. Ther. 2012, 37, 175–184. [Google Scholar]
- Tait, A.R.; Straus, S.K. Phosphorylation of U24 from Human Herpes Virus type 6 (HHV-6) and its potential role in mimicking myelin basic protein (MBP) in multiple sclerosis. FEBS Lett. 2008, 582, 2685–2688. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gh Popescu, B.F.; Lucchinetti, C.F. Meningeal and cortical grey matter pathology in multiple sclerosis. BMC Neurol. 2012, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Rosati, G. The prevalence of multiple sclerosis in the world: An update. Neurol. Sci. 2001, 22, 117–139. [Google Scholar] [CrossRef]
- Abbaszadeh, S.; Tabary, M.; Aryannejad, A.; Abolhasani, R.; Araghi, F.; Khaheshi, I.; Azimi, A. Air pollution and multiple sclerosis: A comprehensive review. Neurol. Sci. 2021, 42, 4063–4072. [Google Scholar] [CrossRef]
- Rimkus, C.M.; Schoeps, V.A.; Boaventura, M.; Godoy, L.F.; Apostolos-Pereira, S.L.; Calich, A.L.; Callegaro, D.; Lucato, L.T.; Rovira, A.; Sastre-Garriga, J.; et al. Drug-related demyelinating syndromes: Understanding risk factors, pathophysiological mechanisms and magnetic resonance imaging findings. Mult. Scler. Relat. Disord. 2021, 55, 103146. [Google Scholar] [CrossRef]
- Obermeier, B.; Mentele, R.; Malotka, J.; Kellermann, J.; Kümpfel, T.; Wekerle, H.; Lottspeich, F.; Hohlfeld, R.; Dornmair, K. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat. Med. 2008, 14, 688–693. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef]
- Holloman, J.P.; Axtell, R.C.; Monson, N.L.; Wu, G.F. The Role of B Cells in Primary Progressive Multiple Sclerosis. Front. Neurol. 2021, 12, 877. [Google Scholar] [CrossRef]
- Leibovitch, E.C.; Jacobson, S. Evidence linking HHV-6 with multiple sclerosis: An update. Curr. Opin. Virol. 2014, 9, 127–133. [Google Scholar] [CrossRef]
- Wucherpfennig, K.W.; Strominger, J.L. Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995, 80, 695–705. [Google Scholar] [CrossRef]
- Veroni, C.; Aloisi, F. The CD8 T Cell-Epstein-Barr Virus-B Cell Trialogue: A Central Issue in Multiple Sclerosis Pathogenesis. Front. Immunol. 2021, 12, 2450. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, M.; Martin, R. Immunology of Multiple Sclerosis. Semin. Neurol. 2016, 36, 115–127. [Google Scholar] [CrossRef]
- Lincoln, M.R.; Montpetit, A.; Cader, M.Z.; Saarela, J.; Dyment, D.A.; Tiislar, M.; Ferretti, V.; Tienari, P.J.; Sadovnick, A.D.; Peltonen, L.; et al. A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat. Genet. 2005, 37, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Goris, A.; Pauwels, I.; Dubois, B. Progress in Multiple Sclerosis Genetics. Curr. Genom. 2012, 13, 646–663. [Google Scholar] [CrossRef]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [PubMed]
- Patsopoulos, N.A.; Baranzini, S.E.; Santaniello, A.; Shoostari, P.; Cotsapas, C.; Wong, G.; Beecham, A.H.; James, T.; Replogle, J.; Vlachos, I.S.; et al. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1362. [Google Scholar] [CrossRef]
- Maglione, A.; Zuccalà, M.; Tosi, M.; Clerico, M.; Rolla, S. Host genetics and gut microbiome: Perspectives for multiple sclerosis. Genes 2021, 12, 1181. [Google Scholar] [CrossRef]
- Waubant, E.; Lucas, R.; Mowry, E.; Graves, J.; Olsson, T.; Alfredsson, L.; Langer-Gould, A. Environmental and genetic risk factors for MS: An integrated review. Ann. Clin. Transl. Neurol. 2019, 6, 1905–1922. [Google Scholar] [CrossRef]
- Schwartz, K.L.; Richardson, S.E.; Ward, K.N.; Donaldson, C.; MacGregor, D.; Banwell, B.; Mahant, S.; Bitnun, A. Delayed Primary HHV-7 Infection and Neurologic Disease. Pediatrics 2014, 133, e1541-7. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ma, D.; Li, L.; Zhang, L. Progress in the Application of Drugs for the Treatment of Multiple Sclerosis. Front. Pharmacol. 2021, 12, 1793. [Google Scholar] [CrossRef] [PubMed]
- Fotheringham, J.; Jacobson, S. Human herpesvirus 6 and multiple sclerosis: Potential mechanisms for virus-induced disease. Herpes 2005, 12, 4–9. [Google Scholar] [PubMed]
- Christensen, T. Human herpesviruses in MS. Int. MS J. 2007, 14, 41–47. [Google Scholar] [PubMed]
- Bello-Morales, R.; Andreu, S.; Ripa, I.; López-Guerrero, J.A. Hsv-1 and endogenous retroviruses as risk factors in demyelination. Int. J. Mol. Sci. 2021, 22, 5738. [Google Scholar] [CrossRef] [PubMed]
- Levin, L.I.; Munger, K.L.; O’Reilly, E.J.; Falk, K.I.; Ascherio, A. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann. Neurol. 2010, 67, 824–830. [Google Scholar] [CrossRef]
- Levin, L.I.; Munger, K.L.; Rubertone, M.V.; Peck, C.A.; Lennette, E.T.; Spiegelman, D.; Ascherio, A. Temporal relationship between elevation of Epstein-Barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. J. Am. Med. Assoc. 2005, 293, 2496–2500. [Google Scholar] [CrossRef]
- Sundström, P.; Juto, P.; Wadell, G.; Hallmans, G.; Svenningsson, A.; Nyström, L.; Dillner, J.; Forsgren, L. An altered immune response to Epstein-Barr virus in multiple sclerosis: A prospective study. Neurology 2004, 62, 2277–2282. [Google Scholar] [CrossRef]
- DeLorenze, G.N.; Munger, K.L.; Lennette, E.T.; Orentreich, N.; Vogelman, J.H.; Ascherio, A. Epstein-Barr virus and multiple sclerosis: Evidence of association from a prospective study with long-term follow-up. Arch. Neurol. 2006, 63, 839–844. [Google Scholar] [CrossRef]
- Höllsberg, P.; Hansen, H.J.; Haahr, S. Altered CD8+ T cell responses to selected Epstein-Barr virus immunodominant epitopes in patients with multiple sclerosis. Clin. Exp. Immunol. 2003, 132, 137–143. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Edwards, N.; Muraro, P.A.; Hayashi, S.; Cohen, J.I.; Münz, C.; Martin, R. Increased frequency and broadened specificity of latent EBV nuclear antigen-1-specific T cells in multiple sclerosis. Brain 2006, 129, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Jilek, S.; Schluep, M.; Meylan, P.; Vingerhoets, F.; Guignard, L.; Monney, A.; Kleeberg, J.; Le Goff, G.; Pantaleo, G.; Du Pasquier, R.A. Strong EBV-specific CD8+ T-cell response in patients with early multiple sclerosis. Brain 2008, 131, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Latham, L.B.; Lee, M.J.; Lincoln, J.A.; Ji, N.; Forsthuber, T.G.; Lindsey, J.W. Antivirus immune activity in multiple sclerosis correlates with MRI activity. Acta Neurol. Scand. 2016, 133, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Pender, M.P.; Csurhes, P.A.; Burrows, J.M.; Burrows, S.R. Defective T-cell control of Epstein-Barr virus infection in multiple sclerosis. Clin. Transl. Immunol. 2017, 6, e126. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Veroni, C.; Mazzola, G.A.; Aloisi, F. Epstein-Barr Virus-Specific CD8 T Cells Selectively Infiltrate the Brain in Multiple Sclerosis and Interact Locally with Virus-Infected Cells: Clue for a Virus-Driven Immunopathological Mechanism. J. Virol. 2019, 93, e00980-19. [Google Scholar] [CrossRef]
- Jog, N.R.; McClain, M.T.; Heinlen, L.D.; Gross, T.; Towner, R.; Guthridge, J.M.; Axtell, R.C.; Pardo, G.; Harley, J.B.; James, J.A. Epstein Barr Virus Nuclear Antigen 1 (EBNA-1) peptides recognized by adult multiple sclerosis patient sera induce neurologic symptoms in a murine model. J. Autoimmun. 2020, 106, 102332. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, Y.; Arapidi, G.P.; Chernov, A.; Ziganshin, R.; Tcyganov, E.; Lyadova, I.; Butenko, I.O.; Osetrova, M.; Ponomarenko, N.; Telegin, G.; et al. Exposure to the Epstein-Barr viral antigen latent membrane protein 1 induces myelin-reactive antibodies in vivo. Front. Immunol. 2017, 8, 777. [Google Scholar] [CrossRef] [PubMed]
- Van Noort, J.M.; Bajramovic, J.J.; Plomp, A.C.; Van Stipdonk, M.J.B. Mistaken self, a novel model that links microbial infections with myelin-directed autoimmunity in multiple sclerosis. J. Neuroimmunol. 2000, 105, 46–57. [Google Scholar] [CrossRef]
- Guo, Y.S.; Liang, P.Z.; Lu, S.Z.; Chen, R.; Yin, Y.Q.; Zhou, J.W. Extracellular αB-crystallin modulates the inflammatory responses. Biochem. Biophys. Res. Commun. 2019, 508, 282–288. [Google Scholar] [CrossRef]
- Geginat, J.; Paroni, M.; Pagani, M.; Galimberti, D.; De Francesco, R.; Scarpini, E.; Abrignani, S. The Enigmatic Role of Viruses in Multiple Sclerosis: Molecular Mimicry or Disturbed Immune Surveillance? Trends Immunol. 2017, 38, 498–512. [Google Scholar] [CrossRef]
- Harauz, G.; Boggs, J.M. Myelin management by the 18.5-kDa and 21.5-kDa classic myelin basic protein isoforms. J. Neurochem. 2013, 125, 334–361. [Google Scholar] [CrossRef]
- Panitch, H.S.; Hooper, C.J.; Johnson, K.P. CSF Antibody to Myelin Basic Protein. Arch. Neurol. 1980, 37, 206. [Google Scholar] [CrossRef] [PubMed]
- Reindl, M.; Linington, C.; Brehm, U.; Egg, R.; Dilitz, E.; Deisenhammer, F.; Poewe, W.; Berger, T. Antibodies against the myelin oligodendrocyte glycoprotein and the myelin basic protein in multiple sclerosis and other neurological diseases: A comparative study. Brain 1999, 122, 2047–2056. [Google Scholar] [CrossRef] [PubMed]
- Hegen, H.; Reindl, M. Recent developments in MOG-IgG associated neurological disorders. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420945135. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Mastronardi, F.G.; Wood, D.D.; Lubman, D.M.; Zand, R.; Moscarello, M.A. Multiple sclerosis: An important role for post-translational modifications of myelin basic protein in pathogenesis. Mol. Cell. Proteom. 2003, 2, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Longnecker, R.; Merchant, M.; Brown, M.E.; Fruehling, S.; Bickford, J.O.; Ikeda, M.; Harty, R.N. WW- and SH3-domain interactions with epstein-barr virus LMP2A. Exp. Cell Res. 2000, 257, 332–340. [Google Scholar] [CrossRef]
- Winberg, G.; Matskova, L.; Chen, F.; Plant, P.; Rotin, D.; Gish, G.; Ingham, R.; Ernberg, I.; Pawson, T. Latent Membrane Protein 2A of Epstein-Barr Virus Binds WW Domain E3 Protein-Ubiquitin Ligases That Ubiquitinate B-Cell Tyrosine Kinases. Mol. Cell. Biol. 2000, 20, 8526–8535. [Google Scholar] [CrossRef] [PubMed]
- Panousis, C.G.; Rowe, D.T. Epstein-Barr virus latent membrane protein 2 associates with and is a substrate for mitogen-activated protein kinase. J. Virol. 1997, 71, 4752–4760. [Google Scholar] [CrossRef]
- Kearns, P.K.A.; Casey, H.A.; Leach, J.P. Hypothesis: Multiple sclerosis is caused by three-hits, strictly in order, in genetically susceptible persons. Mult. Scler. Relat. Disord. 2018, 24, 157–174. [Google Scholar] [CrossRef]
- Aloisi, F.; Serafini, B.; Magliozzi, R.; Howell, O.W.; Reynolds, R. Detection of Epstein-Barr virus and B-cell follicles in the multiple sclerosis brain: What you find depends on how and where you look. Brain 2010, 133, e157. [Google Scholar] [CrossRef]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [PubMed]
- Pender, M.P.; Csurhes, P.A.; Smith, C.; Beagley, L.; Hooper, K.D.; Raj, M.; Coulthard, A.; Burrows, S.R.; Khanna, R. Epstein-Barr virus-specific adoptive immunotherapy for progressive multiple sclerosis. Mult. Scler. J. 2014, 20, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, S.B.; Obeidat, A.Z. EBNA1, EBNA2, and EBNA3 link Epstein-Barr virus and hypovitaminosis D in multiple sclerosis pathogenesis. J. Neuroimmunol. 2020, 339, 577116. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.L.; Levin, L.I.; O’Reilly, E.J.; Falk, K.I.; Ascherio, A. Anti-Epstein-Barr virus antibodies as serological markers of multiple sclerosis: A prospective study among United States military personnel. Mult. Scler. J. 2011, 17, 1185–1193. [Google Scholar] [CrossRef]
- Kaiser, C.; Laux, G.; Eick, D.; Jochner, N.; Bornkamm, G.W.; Kempkes, B. The Proto-Oncogene c- myc Is a Direct Target Gene of Epstein-Barr Virus Nuclear Antigen 2. J. Virol. 1999, 73, 4481–4484. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W.; et al. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 2010, 8, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. A region of herpes simplex virus VP16 can substitute for a transforming domain of Epstein-Barr virus nuclear protein 2. Proc. Natl. Acad. Sci. USA 1992, 89, 8030–8034. [Google Scholar] [CrossRef]
- Wang, L.; Grossman, S.R.; Kieff, E. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 430–435. [Google Scholar] [CrossRef]
- Zhao, B.; Zou, J.; Wang, H.; Johannsen, E.; Peng, C.W.; Quackenbush, J.; Mar, J.C.; Morton, C.C.; Freedman, M.L.; Blacklow, S.C.; et al. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 14902–14907. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Franciotta, D.; Magliozzi, R.; Reynolds, R.; Cinque, P.; Andreoni, L.; Trivedi, P.; Salvetti, M.; Faggioni, A.; et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J. Exp. Med. 2007, 204, 2899–2912. [Google Scholar] [CrossRef]
- Challoner, P.B.; Smith, K.T.; Parker, J.D.; MacLeod, D.L.; Coulter, S.N.; Rose, T.M.; Schultz, E.R.; Bennett, J.L.; Garber, R.L.; Chang, M. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc. Natl. Acad. Sci. USA 1995, 92, 7440–7444. [Google Scholar] [CrossRef] [PubMed]
- Caserta, M.T.; Hall, C.B.; Schnabel, K.; McIntyre, K.; Long, C.; Costanzo, M.; Dewhurst, S.; Insel, R.; Epstein, L.G. Neuroinvasion and persistence of human herpesvirus 6 in children. J. Infect. Dis. 1994, 170, 1586–1589. [Google Scholar] [CrossRef] [PubMed]
- Sola, P.; Merelli, E.; Marasca, R.; Poggi, M.; Luppi, M.; Montorsi, M.; Torelli, G. Human herpesvirus 6 and multiple sclerosis: Survey of anti-HHV-6 antibodies by immunofluorescence analysis and of viral sequences by polymerase chain reaction. J. Neurol. Neurosurg. Psychiatry 1993, 56, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, S.Z.; Ablashi, D.V.; Markham, P.D.; Josephs, S.F.; Sturzenegger, S.; Kaplan, M.; Halligan, G.; Biberfeld, P.; Wong-Staal, F.; Kramarsky, B.; et al. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 1986, 234, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Leibovitch, E.C.; Jacobson, S. Human Herpesvirus 6 as a Viral Trigger in Mesial Temporal Lobe Epilepsy. J. Infect. Dis. 2015, 212, 1011–1013. [Google Scholar] [CrossRef]
- Theodore, W.H.; Leibovitch, E.; Billioux, B.J.; Inati, S.K.; Zaghloul, K.; Heiss, J.; Gaillard, W.D.; Jacobson, S. Human herpesvirus 6 and epilepsy. Epilepsia Open 2021, 6, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; Bortolotti, D.; Gentili, V.; Rotola, A.; Bolzani, S.; Caselli, E.; Tola, M.R.; Di Luca, D. KIR2DS2/KIR2DL2/HLA-C1 Haplotype Is Associated with Alzheimer’s Disease: Implication for the Role of Herpesvirus Infections. J. Alzheimer’s Dis. 2019, 67, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Allnutt, M.A.; Johnson, K.; Bennett, D.A.; Connor, S.M.; Troncoso, J.C.; Pletnikova, O.; Albert, M.S.; Resnick, S.M.; Scholz, S.W.; De Jager, P.L.; et al. Human Herpesvirus 6 Detection in Alzheimer’s Disease Cases and Controls across Multiple Cohorts. Neuron 2020, 105, 1027–1035.e2. [Google Scholar] [CrossRef]
- Schirmer, E.C.; Wyatt, L.S.; Yamanishi, K.; Rodriguez, W.J.; Frenkel, N. Differentiation between two distinct classes of viruses now classified as human herpesvirus 6. Proc. Natl Acad. Sci. USA 1991, 88, 5922–5926. [Google Scholar] [CrossRef]
- Isegawa, Y.; Mukai, T.; Nakano, K.; Kagawa, M.; Chen, J.; Mori, Y.; Sunagawa, T.; Kawanishi, K.; Sashihara, J.; Hata, A.; et al. Comparison of the complete DNA sequences of human herpesvirus 6 variants A and B. J. Virol. 1999, 73, 8053–8063. [Google Scholar] [CrossRef]
- Cohrs, R.J.; Gilden, D.H. Human herpesvirus latency. Brain Pathol. 2001, 11, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Slobedman, B.; Mocarski, E.S. Quantitative analysis of latent human cytomegalovirus. J. Virol. 1999, 73, 4806–4812. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kondo, T.; Okuno, T.; Takahashi, M.; Ymanishi, K. Latent human herpesvirus 6 infection of human monocytes/macrophages. J. Gen. Virol. 1991, 72, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Shimada, K.; Sashihara, J.; Tanaka-Taya, K.; Yamanishi, K. Identification of Human Herpesvirus 6 Latency-Associated Transcripts. J. Virol. 2002, 76, 4145–4151. [Google Scholar] [CrossRef]
- Gordon, L.; McQuaid, S.; Cosby, S.L. Detection of herpes simplex virus (types 1 and 2) and human herpesvirus 6 DNA in human brain tissue by polymerase chain reaction. Clin. Diagn. Virol. 1996, 6, 33–40. [Google Scholar] [CrossRef]
- Merelli, E.; Bedin, R.; Sola, P.; Barozzi, P.; Mancardi, G.L.; Ficarra, G.; Franchini, G. Human herpes virus 6 and human herpes virus 8 DNA sequences in brains of multiple sclerosis patients, normal adults and children. J. Neurol. 1997, 244, 450–454. [Google Scholar] [CrossRef]
- Cermelli, C.; Berti, R.; Soldan, S.S.; Mayne, M.; D’Ambrosia, J.M.; Ludwin, S.K.; Jacobson, S. High frequency of human herpesvirus 6 DNA in multiple sclerosis plaques isolated by laser microdissection. J. Infect. Dis. 2003, 187, 1377–1387. [Google Scholar] [CrossRef]
- Sanders, V.J.; Felisan, S.; Waddell, A.; Tourtellotte, W.W. Detection of Herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J. Neurovirol. 1996, 2, 249–258. [Google Scholar] [CrossRef]
- Opsahl, M.L.; Kennedy, P.G. Early and late HHV-6 gene transcripts in multiple sclerosis lesions and normal appearing white matter. Brain 2005, 128, 516–527. [Google Scholar] [CrossRef]
- Leibovitch, E.C.; Caruso, B.; Ha, S.K.; Schindler, M.K.; Lee, N.J.; Luciano, N.J.; Billioux, B.J.; Guy, J.R.; Yen, C.; Sati, P.; et al. Herpesvirus trigger accelerates neuroinflammation in a nonhuman primate model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 11292–11297. [Google Scholar] [CrossRef]
- Tejada-Simon, M.V.; Zang, Y.C.Q.; Hong, J.; Rivera, V.M.; Zhang, J.Z. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann. Neurol. 2003, 53, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, A.; Johnson, R.T. Infections and multiple sclerosis. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2014; Volume 122, pp. 151–171. [Google Scholar]
- Owens, G.P.; Gilden, D.; Burgoon, M.P.; Yu, X.; Bennett, J.L. Viruses and multiple sclerosis. Neuroscientist 2011, 17, 659–676. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Ma, Y.; Gong, F.; Hu, C.; Qian, L.; Huang, Q.; Yu, Q.; Zhang, J.; Chen, S.; Liu, Z.; et al. Cross-reactivity of autoreactive T cells with MBP and viral antigens in patients with MS. Front. Biosci. 2012, 17, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; D’Accolti, M.; Caccuri, F.; Soffritti, I.; Gentili, V.; Bortolotti, D.; Rotola, A.; Cassai, E.; Fiorentini, S.; Zani, A.; et al. The U94 Gene of Human Herpesvirus 6: A Narrative Review of Its Role and Potential Functions. Cells 2020, 9, 2608. [Google Scholar] [CrossRef] [PubMed]
- Ben-Fredj, N.; Ben-Selma, W.; Rotola, A.; Nefzi, F.; Benedetti, S.; Frih-Ayed, M.; Di Luca, D.; Aouni, M.; Caselli, E. Prevalence of human herpesvirus U94/REP antibodies and DNA in Tunisian multiple sclerosis patients. J. Neurovirol. 2013, 19, 42–47. [Google Scholar] [CrossRef]
- Campbell, A.; Hogestyn, J.M.; Folts, C.J.; Lopez, B.; Pröschel, C.; Mock, D.; Mayer-Pröschel, M. Expression of the Human Herpesvirus 6A Latency-Associated Transcript U94A Disrupts Human Oligodendrocyte Progenitor Migration. Sci. Rep. 2017, 7, 3978. [Google Scholar] [CrossRef]
- Thakolwiboon, S.; Zhao-Fleming, H.; Karukote, A.; Pachariyanon, P.; Williams, H.G.; Avila, M. Regional differences in the association of cytomegalovirus seropositivity and multiple sclerosis: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2020, 45, 102393. [Google Scholar] [CrossRef]
- Langer-Gould, A.; Wu, J.; Lucas, R.; Smith, J.; Gonzales, E.; Amezcua, L.; Haraszti, S.; Chen, L.H.; Quach, H.; James, J.A.; et al. Epstein-Barr virus, cytomegalovirus, and multiple sclerosis susceptibility. Neurology 2017, 89, 1330–1337. [Google Scholar] [CrossRef]
- Do Olival, G.S.; Lima, B.M.; Sumita, L.M.; Serafim, V.; Fink, M.C.; Nali, L.H.; Romano, C.M.; Thomaz, R.B.; Cavenaghi, V.B.; Tilbery, C.P.; et al. Multiple sclerosis and herpesvirus interaction. Arq. Neuropsiquiatr. 2013, 71, 727–730. [Google Scholar] [CrossRef]
- Nobre, L.; Nightingale, K.; Ravenhill, B.J.; Antrobus, R.; Soday, L.; Nichols, J.; Davies, J.; Seirafian, S.; Wang, E.C.Y.; Davison, A.J.; et al. Human cytomegalovirus interactome analysis identifies degradation hubs, domain associations and viral protein functions. eLife 2019, 8, e49894. [Google Scholar] [CrossRef]
- Koshizuka, T.; Kobayashi, T.; Ishioka, K.; Suzutani, T. Herpesviruses possess conserved proteins for interaction with Nedd4 family ubiquitin E3 ligases. Sci. Rep. 2018, 8, 4447. [Google Scholar] [CrossRef] [PubMed]
- Koshizuka, T.; Kondo, H.; Kato, H.; Takahashi, K. Human cytomegalovirus UL42 protein inhibits the degradation of glycoprotein B through inhibition of Nedd4 family ubiquitin E3 ligases. Microbiol. Immunol. 2021, 65, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.; Mogensen, T.H. Determinants of neurological syndromes caused by varicella zoster virus (VZV). J. Neurovirol. 2020, 26, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Manouchehrinia, A.; Tanasescu, R.; Kareem, H.; Jerca, O.P.; Jabeen, F.; Shafei, R.; Breuer, J.; Neal, K.; Irving, W.; Constantinescu, C.S. Prevalence of a history of prior varicella/herpes zoster infection in multiple sclerosis. J. Neurovirol. 2017, 23, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Ordoñez, G.; Pineda, B.; Flores, J. The participation of varicella zoster virus in relapses of multiple sclerosis. Clin. Neurol. Neurosurg. 2014, 119, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Burgoon, M.P.; Cohrs, R.J.; Bennett, J.L.; Anderson, S.W.; Ritchie, A.M.; Cepok, S.; Hemmer, B.; Gilden, D.; Owens, G.P. Varicella zoster virus is not a disease-relevant antigen in multiple sclerosis. Ann. Neurol. 2009, 65, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Czarnowska, A.; Kapica-Topczewska, K.; Zajkowska, O.; Świerzbińska, R.; Chorąży, M.; Tarasiuk, J.; Zajkowska, J.; Kochanowicz, J.; Kułakowska, A. Herpesviridae Seropositivity in Patients with Multiple Sclerosis: First Polish Study. Eur. Neurol. 2018, 80, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Geny, C.; Laurent, A.; Mouriquand, C.; Pellat, J.; Perret, J.; Seigneurin, J.M. Leptomeningeal cell line from multiple sclerosis with reverse transcriptase activity and viral particles. Res. Virol. 1989, 140, 551–561. [Google Scholar] [CrossRef]
- Arneth, B. Up-to-date knowledge about the association between multiple sclerosis and the reactivation of human endogenous retrovirus infections. J. Neurol. 2018, 265, 1733–1739. [Google Scholar] [CrossRef]
- Küry, P.; Nath, A.; Créange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef]
- Morandi, E.; Tanasescu, R.; Tarlinton, R.E.; Constantinescu, C.S.; Zhang, W.; Tench, C.; Gran, B. The association between human endogenous retroviruses and multiple sclerosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0172415. [Google Scholar] [CrossRef] [PubMed]
- Dolei, A. The aliens inside us: HERV-W endogenous retroviruses and multiple sclerosis. Mult. Scler. 2018, 24, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Jakhmola, S.; Upadhyay, A.; Jain, K.; Mishra, A.; Jha, H.C. Herpesviruses and the hidden links to Multiple Sclerosis neuropathology. J. Neuroimmunol. 2021, 358, 577636. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Bernard, C.; Bertrand, J.B.; Lang, A.B.; Popa, I.; Sanhadji, K.; Portoukalian, J. Endogenous retroviral genes, Herpesviruses and gender in Multiple Sclerosis. J. Neurol. Sci. 2009, 286, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Van Der Pol, S.; Nijland, P.; Amor, S.; Perron, H. Human endogenous retrovirus W in brain lesions: Rationale for targeted therapy in multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 8, 11–18. [Google Scholar] [CrossRef]
- Johnston, J.B.; Silva, C.; Holden, J.; Warren, K.G.; Clark, A.W.; Power, C. Monocyte activation and differentiation augment human endogenous retrovirus expression: Implications for inflammatory brain diseases. Ann. Neurol. 2001, 50, 434–442. [Google Scholar] [CrossRef]
- Perron, H.; Lazarini, F.; Ruprecht, K.; Péchoux-Longin, C.; Seilhean, D.; Sazdovitch, V.; Créange, A.; Battail-Poirot, N.; Sibaï, G.; Santoro, L.; et al. Human endogenous retrovirus (HERV)-W ENV and GAG proteins: Physiological expression in human brain and pathophysiological modulation in multiple sclerosis lesions. J. Neurovirol. 2005, 11, 23–33. [Google Scholar] [CrossRef]
- Mameli, G.; Astone, V.; Arru, G.; Marconi, S.; Lovato, L.; Serra, C.; Sotgiu, S.; Bonetti, B.; Dolei, A. Brains and peripheral blood mononuclear cells of multiple sclerosis (MS) patients hyperexpress MS-associated retrovirus/HERV-W endogenous retrovirus, but not human herpesvirus 6. J. Gen. Virol. 2007, 88, 264–274. [Google Scholar] [CrossRef]
- Ayers, K.N.; Carey, S.N.; Lukacher, A.E. Understanding polyomavirus CNS disease—A perspective from mouse models. FEBS J. 2021. [Google Scholar] [CrossRef]
- Cortese, I.; Reich, D.S.; Nath, A. Progressive multifocal leukoencephalopathy and the spectrum of JC virus-related disease. Nat. Rev. Neurol. 2021, 17, 37–51. [Google Scholar] [CrossRef]
- Ho, P.R.; Koendgen, H.; Campbell, N.; Haddock, B.; Richman, S.; Chang, I. Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: A retrospective analysis of data from four clinical studies. Lancet Neurol. 2017, 16, 925–933. [Google Scholar] [CrossRef]
- Komaroff, A.L.; Pellett, P.E.; Jacobson, S. Human herpesviruses 6a and 6b in brain diseases: Association versus causation. Clin. Microbiol. Rev. 2021, 34, e00143-20. [Google Scholar] [CrossRef] [PubMed]
- Brütting, C.; Stangl, G.I.; Staege, M.S. Vitamin D, Epstein-Barr virus, and endogenous retroviruses in multiple sclerosis—facts and hypotheses. J. Integr. Neurosci. 2021, 20, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Zarrinpar, A.; Bhattacharyya, R.P.; Lim, W.A. The structure and function of proline recognition domains. Sci. STKE 2003, 2003, re8. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.J.; Wiesner, S.; Sudol, M. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2002, 513, 30–37. [Google Scholar] [CrossRef]
- Shepley-McTaggart, A.; Fan, H.; Sudol, M.; Harty, R.N.; Cameron, C.E. Viruses go modular. J. Biol. Chem. 2020, 295, 4604–4616. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Mayer, B.J. SH3 domains: Complexity in moderation. J. Cell Sci. 2001, 114, 1253–1263. [Google Scholar] [CrossRef]
- Musacchio, A. How SH3 domains recognize proline. Adv. Protein Chem. 2002, 61, 211–268. [Google Scholar]
- Feng, S.; Chen, J.K.; Yu, H.; Simon, J.A.; Schreiber, S.L. Two binding orientations for peptides to the Src SH3 domain: Development of a general model for SH3-ligand interactions. Science 1994, 266, 1241–1247. [Google Scholar] [CrossRef]
- Feng, S.; Kasahara, C.; Rickles, R.J.; Schreiber, S.L. Specific interactions outside the proline-rich core of two classes of Src homology 3 ligands. Proc. Natl. Acad. Sci. USA 1995, 92, 12408–12415. [Google Scholar] [CrossRef] [PubMed]
- Demers, J.P.; Mittermaier, A. Binding mechanism of an SH3 domain studied by NMR and ITC. J. Am. Chem. Soc. 2009, 131, 4355–4367. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.Y.H.; Nie, J.; Wu, C.; Li, C.; Li, S.S.C. Novel Src homology 3 domain-binding motifs identified from proteomic screen of a pro-rich region. Mol. Cell. Proteom. 2005, 4, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Ibragimova, G.T.; Wade, R.C. Importance of explicit salt ions for protein stability in molecular dynamics simulation. Biophys. J. 1998, 74, 2906–2911. [Google Scholar] [CrossRef]
- Xin, H.; Poy, F.; Zhang, R.; Joachimiak, A.; Sudol, M.; Eck, M.J. Structure of a WW domain containing fragment of dystrophin in complex with β-dystroglycan. Nat. Struct. Biol. 2000, 7, 634–638. [Google Scholar] [CrossRef]
- Staub, O.; Abriel, H.; Plant, P.; Isiiikawa, T.; Kanelis, V.; Saleki, R.; Horisberger, J.D.; Schild, L.; Rotin, D. Regulation of the epithelial Na+ channel by Nedd4 and ubiquitination. Kidney Int. 2000, 57, 809–815. [Google Scholar] [CrossRef]
- Passani, L.A.; Bedford, M.T.; Faber, P.W.; McGinnis, K.M.; Sharp, A.H.; Gusella, J.F.; Vonsattel, J.P.; MacDonald, M.E. Huntingtin’s WW domain partners in Huntington’s disease post-mortem brain fulfill genetic criteria for direct involvement in Huntington’s disease pathogenesis. Hum. Mol. Genet. 2000, 9, 2175–2182. [Google Scholar] [CrossRef]
- Yin, Q.; Wyatt, C.J.; Han, T.; Smalley, K.S.M.; Wan, L. ITCH as a potential therapeutic target in human cancers. Semin. Cancer Biol. 2020, 67, 117–130. [Google Scholar] [CrossRef]
- Ilsley, J.L.; Sudol, M.; Winder, S.J. The WW domain: Linking cell signalling to the membrane cytoskeleton. Cell. Signal. 2002, 14, 183–189. [Google Scholar] [CrossRef]
- Hu, H.; Columbus, J.; Zhang, Y.; Wu, D.; Lian, L.; Yang, S.; Goodwin, J.; Luczak, C.; Carter, M.; Chen, L.; et al. A map of WW domain family interactions. Proteomics 2004, 4, 643–655. [Google Scholar] [CrossRef]
- Vassall, K.A.; Bamm, V.V.; Harauz, G. MyelStones: The executive roles of myelin basic protein in myelin assembly and destabilization in multiple sclerosis. Biochem. J. 2015, 472, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Guglietti, B.; Sivasankar, S.; Mustafa, S.; Corrigan, F.; Collins-Praino, L.E. Fyn Kinase Activity and Its Role in Neurodegenerative Disease Pathology: A Potential Universal Target? Mol. Neurobiol. 2021, 58, 5986–6005. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Petrillo, F.; Nasso, R.; Ferretti, G. Fyn tyrosine kinase as harmonizing factor in neuronal functions and dysfunctions. Int. J. Mol. Sci. 2020, 21, 4444. [Google Scholar] [CrossRef] [PubMed]
- Gonsior, C.; Binamé, F.; Frühbeis, C.; Bauer, N.M.; Hoch-Kraft, P.; Luhmann, H.J.; Trotter, J.; White, R. Oligodendroglial p130Cas is a target of Fyn kinase involved in process formation, cell migration and survival. PLoS ONE 2014, 9, e89423. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Ku, L.; Chen, Y.; Feng, Y. Developmental abnormalities of myelin basic protein expression in fyn knock-out brain reveal a role of fyn in posttranscriptional regulation. J. Biol. Chem. 2005, 280, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Umemori, H.; Kadowaki, Y.; Hirosawa, K.; Yoshida, Y.; Hironaka, K.; Okano, H.; Yamamoto, T. Stimulation of myelin basic protein gene transcription by fyn tyrosine kinase for myelination. J. Neurosci. 1999, 19, 1393–1397. [Google Scholar] [CrossRef][Green Version]
- Polverini, E.; Rangaraj, G.; Libich, D.S.; Boggs, J.M.; Harauz, G. Binding of the proline-rich segment of myelin basic protein to SH3 domains: Spectroscopic, microarray, and modeling studies of ligand conformation and effects of posttranslational modifications. Biochemistry 2008, 47, 267–282. [Google Scholar] [CrossRef]
- Smith, G.S.T.; Homchaudhuri, L.; Boggs, J.M.; Harauz, G. Classic 18.5- and 21.5-kDa myelin basic protein isoforms associate with cytoskeletal and SH3-domain proteins in the immortalized N19-oligodendroglial cell line stimulated by phorbol ester and IGF-1. Neurochem. Res. 2012, 37, 1277–1295. [Google Scholar] [CrossRef][Green Version]
- Smith, G.S.T.; De Avila, M.; Paez, P.M.; Spreuer, V.; Wills, M.K.B.; Jones, N.; Boggs, J.M.; Harauz, G. Proline substitutions and threonine pseudophosphorylation of the SH3 ligand of 18.5-kDa myelin basic protein decrease its affinity for the Fyn-SH3 domain and alter process development and protein localization in oligodendrocytes. J. Neurosci. Res. 2012, 90, 28–47. [Google Scholar] [CrossRef]
- White, R.; Krämer-Albers, E.M. Axon-glia interaction and membrane traffic in myelin formation. Front. Cell. Neurosci. 2014, 7, 284. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Iannuzzi, F.; Annunziato, L. The Y682ENPTY687 motif of APP: Progress and insights toward a targeted therapy for Alzheimer’s disease patients. Ageing Res. Rev. 2019, 52, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Schenone, S.; Brullo, C.; Musumeci, F.; Biava, M.; Falchi, F.; Botta, M. Fyn Kinase in Brain Diseases and Cancer: The Search for Inhibitors. Curr. Med. Chem. 2011, 18, 2921–2942. [Google Scholar] [CrossRef] [PubMed]
- Cen, O.; Longnecker, R. Latent membrane protein 2 (LMP2). In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 391, pp. 151–180. [Google Scholar]
- Sang, Y.; Tait, A.R.; Scott, W.R.P.; Creagh, A.L.; Kumar, P.; Haynes, C.A.; Straus, S.K. Probing the interaction between U24 and the SH3 domain of Fyn tyrosine kinase. Biochemistry 2014, 53, 6092–6102. [Google Scholar] [CrossRef] [PubMed]
- De Avila, M.; Vassall, K.A.; Smith, G.S.T.; Bamm, V.V.; Harauz, G. The proline-rich region of 18.5 kDa myelin basic protein binds to the SH3-domain of Fyn tyrosine kinase with the aid of an upstream segment to form a dynamic complex in vitro. Biosci. Rep. 2014, 34, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Kakalacheva, K.; Münz, C.; Lünemann, J.D. Viral triggers of multiple sclerosis. Biochim. Biophys. Acta—Mol. Basis Dis. 2011, 1812, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Jo, J.; Wang, C.Y.; Cristobal, C.D.; Zuo, Z.; Ye, Q.; Wirianto, M.; Lindeke-Myers, A.; Choi, J.M.; Mohila, C.A.; et al. The Daam2-VHL-Nedd4 axis governs developmental and regenerative oligodendrocyte differentiation. Genes Dev. 2020, 34, 1177–1189. [Google Scholar] [CrossRef]
- Boase, N.; Kumar, S. NEDD4: The founding member of a family of ubiquitin-protein ligases. Gene 2015, 557, 113–122. [Google Scholar] [CrossRef]
- Sang, Y.; Zhang, R.; Scott, W.R.P.; Creagh, A.L.; Haynes, C.A.; Straus, S.K. U24 from Roseolovirus interacts strongly with Nedd4 WW Domains. Sci. Rep. 2017, 7, 39776. [Google Scholar] [CrossRef]
- Boggs, J.M.; Rangaraj, G.; Gao, W.; Heng, Y.M. Effect of phosphorylation of myelin basic protein by MAPK on its interactions with actin and actin binding to a lipid membrane in vitro. Biochemistry 2006, 45, 391–401. [Google Scholar] [CrossRef]
- Vassall, K.A.; Bamm, V.V.; Jenkins, A.D.; Velte, C.J.; Kattnig, D.R.; Boggs, J.M.; Hinderberger, D.; Harauz, G. Substitutions mimicking deimination and phosphorylation of 18.5-kDa myelin basic protein exert local structural effects that subtly influence its global folding. Biochim. Biophys. Acta—Biomembr. 2016, 1858, 1262–1277. [Google Scholar] [CrossRef] [PubMed]
- Harauz, G.; Musse, A.A. A tale of two citrullines—Structural and functional aspects of myelin basic protein deimination in health and disease. Neurochem. Res. 2007, 32, 137–158. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.M.; Yon, M.; Groome, N.P.; Sweatt, J.D. Regulation of myelin basic protein phosphorylation by mitogen-activated protein kinase during increased action potential firing in the hippocampus. J. Neurochem. 1999, 73, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.M.; Coscoy, L. The U24 protein from human herpesvirus 6 and 7 affects endocytic recycling. J. Virol. 2010, 84, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Yon, M.; Ackerley, C.A.; Mastronardi, F.G.; Groome, N.; Moscarello, M.A. Identification of a mitogen-activated protein kinase site in human myelin basic protein in situ. J. Neuroimmunol. 1996, 65, 55–59. [Google Scholar] [CrossRef]
- Zuchero, J.B.; Fu, M.M.; Sloan, S.A.; Ibrahim, A.; Olson, A.; Zaremba, A.; Dugas, J.C.; Wienbar, S.; Caprariello, A.V.; Kantor, C.; et al. CNS Myelin Wrapping Is Driven by Actin Disassembly. Dev. Cell 2015, 34, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Krämer-Albers, E.M.; White, R. From axon-glial signalling to myelination: The integrating role of oligodendroglial Fyn kinase. Cell. Mol. Life Sci. 2011, 68, 2003–2012. [Google Scholar] [CrossRef]
- Chang, K.J.; Redmond, S.A.; Chan, J.R. Remodeling myelination: Implications for mechanisms of neural plasticity. Nat. Neurosci. 2016, 19, 190–197. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Source | Protein Name | Segment Sequence | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| myelin | MBP93–107 | I | V | T | P | R | T | P | P | P | S | Q | G | K | G | R | |
| EBV | LMP2A10–105 | G | A | G | P | P | S | P | G | G | D | P | D | G | D | D | G |
| G | N | N | S | Q | Y | P | S | A | S | G | S | S | G | N | T | ||
| P | T | P | P | N | D | E | E | R | E | S | N | E | E | P | P | ||
| P | P | Y | E | D | P | Y | W | G | N | G | D | R | H | S | D | ||
| Y | Q | P | L | G | T | Q | D | Q | S | L | Y | L | G | L | Q | ||
| H | D | G | N | D | G | L | P | P | P | P | Y | S | P | R | D | ||
| HHV-6A | U241–15 | M | D | P | P | R | T | P | P | P | S | Y | S | E | V | L | |
| HHV-6B | U241–15 | M | D | R | P | R | T | P | P | P | S | Y | S | E | V | L | |
| HHV-7 | U241–15 | M | - | T | H | E | T | P | P | P | S | Y | N | D | V | M | L |
| CMV | UL25623–638 | C | R | S | P | P | P | P | L | P | P | R | D | Y | P | Q | R |
| UL4231–46 | S | T | P | P | P | P | P | P | D | C | S | P | P | P | Y | R | |
| VZV | ORF037–52 | A | E | A | V | A | D | A | P | P | P | Y | R | S | R | E | S |
| HSV-1 | UL5615–55 | A | G | N | A | F | A | D | P | P | P | Y | D | S | L | S | G |
| R | N | E | G | P | F | V | V | I | D | L | D | T | P | T | D | ||
| P | P | P | P | Y | S | A | G | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pi, K.-S.; Sang, Y.; Straus, S.K. Viral Proteins with PxxP and PY Motifs May Play a Role in Multiple Sclerosis. Viruses 2022, 14, 281. https://doi.org/10.3390/v14020281
Pi K-S, Sang Y, Straus SK. Viral Proteins with PxxP and PY Motifs May Play a Role in Multiple Sclerosis. Viruses. 2022; 14(2):281. https://doi.org/10.3390/v14020281
Chicago/Turabian StylePi, Keng-Shuo, Yurou Sang, and Suzana K. Straus. 2022. "Viral Proteins with PxxP and PY Motifs May Play a Role in Multiple Sclerosis" Viruses 14, no. 2: 281. https://doi.org/10.3390/v14020281
APA StylePi, K.-S., Sang, Y., & Straus, S. K. (2022). Viral Proteins with PxxP and PY Motifs May Play a Role in Multiple Sclerosis. Viruses, 14(2), 281. https://doi.org/10.3390/v14020281